Búsqueda Simple
Búsqueda Completa
Taxonómica
Anatomía

+ Teoría
Patología
Aleatorio Principio Activo/ComercialFármacos o Drogas Más Visitadas
Levomepromazina

705972 visitas
Clobenzorex
269586 visitas
Desvenlafaxina

266335 visitas
Quetiapina

232325 visitas
Acepromazina
207122 visitas
Últimos comentarios

Sigue las noticias por

Total Fármacos Visitados
14.456.581
Desde Noviembre de 2008
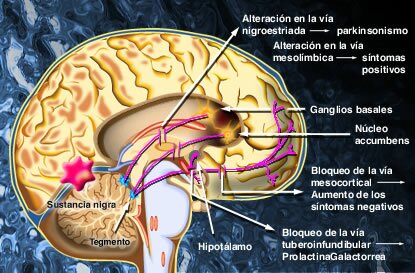
CLASIFICACIÓN DE ANTIPSICÓTICOS


Nombre: Levomepromazina
Nombre Comercial: Sinogan, Nozinan, Togrel
Foto:
Fórmula: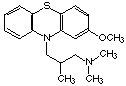
Fármaco consultado 705972 veces
Antipsicóticos Típicos: Levomepromazina
Farmacodinamia También llamados neurolépticos, constituyen un grupo de medicamentos de naturaleza química muy heterogénea pero con mecanismo de acción común. Actúan fundamentalmente por bloqueo de los receptores dopaminérgicos D2, aunque muchos antipsicóticos tienen actividad sobre los receptores de otros neurotrasmisores.
En la actualidad se sabe que la eficacia antipsicótica está estrechamente relacionada con la acción antidopaminérgica a nivel de las vías córtico-meso-límbicas, por bloqueo de los receptores postsinápticos. (4) A su vez la mayoría de efectos adversos neurológicos y endocrinológicos dependen del bloqueo dopaminérgico, aunque no podemos ignorar el protagonismo de diferentes receptores en otros efectos secundarios (sedación por H1 y alfa1, hipotensión ortostática por alfa1, etc.).
Típicos Depot:
Biodisponibilidad de larga duración e inyectados por vía intramuscular, en su mayoría se forman por esterificación de su grupo hidroxilo con un ácido graso de cadena larga; los ésteres se disuelven en distintos tipos de aceite vegetal. Peligrosos por su lenta absorción como su eliminación por el organismo ya que sus metabolitos duran días en el organismo.
Enlace:
Fenotiazinas: Levomepromazina
Se caracterizan por una estructura de 3 anillos (anillos resultantes de la unión de dos anillos benzénicos a través de un puente de N y S. En el grupo de tioxantenos, el puente de N se reemplaza por un puente de C) y difieren entre ellas en las sustituciones realizadas en la posición 2 y 10 (R1 y R2) de la estructura química . En los neurolépticos, derivados fenotiazínicos, la cadena lateral en R1 posee siempre 3 átomos de C seguidos de 1 átomo de N. Esta cadena lateral, es indispensable para el mantenimiento de las propiedades antipsicóticas. La adición de un cloro en R2 origina una asimetría en el núcleo fenotiazínico, con lo que se incrementa la acción farmacológica. El agregado de un radical CF3 en la misma posición, incrementa aún más las acciones antipsicóticas y antieméticas de las fenotiazinas.
Derivados Alifáticos: Levomepromazina
Estas fenotiazinas, tienen dos grupos metilos en el nitrógeno terminal de la cadena lateral (R1) . Poseen una acción sedativa evidente Principalmente antihistamínicos H1. Sus efectos tranquilizantes son intensos por lo que son utilizados comúnmente en episodios esquizofrénicos agudos, excitación maníaca, delirios, agitación ansiosa, etc. Cadena lateral Alifática baja potencia (menor efecto sobre receptores D2), pocos efectos extrapiramidales (3.5%) en comparación con los antipsicóticos convencionales incisivos, producen sedación e hipotensión.
Derivados Piperidínicos:
Son las drogas menos potentes contienen un grupo piperidina, en la cadena lateral. Principalmente antihistamínicos H1. La más conocida es la tioridazina, que tiene una indicación en casos de esquizofrenia con síndromes depresivos . Tomando como base las acciones de la clorpromazina, se ha demostrado que la flufenazina, es a dosis iguales, aproximadamente 20 veces más potente que aquella, y que la trifluoperazina y tioproperazina lo son 10 veces más. La tioridazina, por otra parte, posee solo la mitad de la actividad farmacológica de la clorpromazina. Antipsicóticos de mediana potencia con muy pocos efectos extrapiramidales (0.6%) y con acción sobre receptores 5-HT2
Piperidínicos Depot:
Lo dicho para los típicos depot.
Derivados Piperazínicos:
Son las fenotiazinas más potentes con un grupo piperazina o piperazinil en la cadena lateral. Su acción antipsicótica permite su uso crónico en pacientes esquizofrénicos. Prácticamente no provocan hipotensión ortostática, o su acción es muy pequeña, en este sentido. Son marcadas sin embargo sus acciones extrapiramidales (de mayor intensidad que las dimetílicas). Son activas en dosis menores que las CPZ, y su acción es más rápida provocando escasa acción sedativa. Fármacos más incisivos, con mayores efectos extrapiramidales (7%) y alto riesgo de discinesia tardía. Ventana terapéutica alta. Tienen acción sobre vías nigro-estriales mayor que sobre vías mesolímbicas. Con pocos efectos antimuscarínicos y autonómicos
Piperazínicos Depot:
Lo mismo que lo dicho en los típicos depot.
Tioxantenos:
Derivan del reemplazo del puente de N de las fenotiazinas por un puente de C , manteniéndose la cadena lateral correspondiente. El isómero cis tiene una mayor potencia que el isómero trans. Fármacos de extrapiramidalismo medio (4.1%), con efecto hipotensor, anticolinérgico, sedativo y antiserotoninérgico.
Butirofenonas:
Son compuestos sintéticos. El núcleo butirofenona es una cadena de tres átomos de C unido a un grupo cetónico, y a un anillo bencénico. Todas las butirofenonas, tienen también un átomo de flúor en posición para, del anillo bencénico y la cadena alifática se une a un nitrógeno terciario de un anillo de piperidina (similar a las fenotiazinas). Son potentes agentes neurolépticos de amplio uso psiquiátrico
4-ANILINOPIPERIDÍNICAS:
Caracterizadas por un anillo fenil sustituido. Medicamentos de alta potencia, con mayor efecto extrapiramidal (16%) y ventana terapéutica alta, pero con pocos efectos anticolinérgicos y menor riesgo ocular, hepático y hematológico
Ver gráfico: Correlación entre las dosis clínicamente eficaces de neurolépticos y su capacidad de desplazar al H-haloperidol de su fijación al estriado. (Según Seeman, con autorización de Williamns & Wilkins.)Difenilbutilpiperidinas:
Tienen una gran liposolubilidad y lipofilia, por eso, los lípidos del organismo actúan como depósito de estas drogas, con escasa excreción y larga duración. Están relacionados químicamente con las butirofenonas, con una cadena de tres átomos de C, un anillo piperazínico, y una estructura de dos anillos bencénicos, con un átomo de flúor cada uno en el otro extremo de la cadena
Potentes; causan poca sedación y ninguna hipotensión, pero si un extrapiramidalismo medio
Análogos e isómeros de Fenotiazinas:
Tienen una estructura de tres anillos.
Dibenzoxazepinas:
La posición central lo ocupa la oxazepina, en la estructura tricíclica o heterocíclica
Enlaces:
- (Enciclopédico, en portugués)
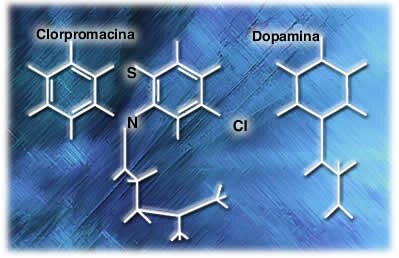
Similitud de estructura química ente la clorpromazina y la dopamina En 1964 los investigadores se dedicaron a buscar una sustancia endógena, químicamente similar a clorpromazina y encontraron dopamina, que aunque tiene una estructura química diferente, sus semejanzas pueden hacerlos activar un mismo receptor. Estudios posteriores demostraron que la clorpromazina se unía a los receptores de dopamina, pero por sus diferencias estructurales no era capaz de activarlo, actuando como antagonista
Enlaces:
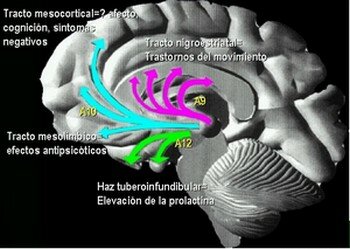
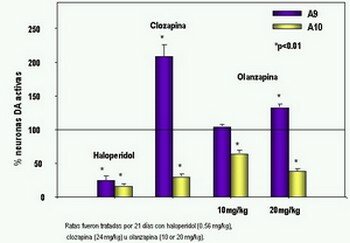
Farmacodinamia Meltzer & Nash clasificaron los antipsicóticos de acuerdo a su afinidad por receptores 5-HT2A v.s. D2, llamando a los que tenían actividad antiserotoninérgica, antipsicóticos atípicos (Meltzer, 1991). Sin embargo, muchos autores han cuestionado que las propiedades atípicas de estos antipsicóticos se deban al bloqueo serotoninérgico, y en cambio, han propuesto que la acción diferencial sobre vías dopaminérgicas A9 (nigroestriatales) y A10 (mesolímbicas) y la afinidad por el receptor D2 sean los responsables de las mismas (Gerlach, 1991 ; Corbett et al., 1993; Benkert & Wetzel, 1994; Vanelle et al., 1994; Seeman et al., 2000). Esto lo hacen partiendo de la observación de que la Risperidona, el cual también ha sido considerado un antipsicótico atípico, a dosis mayores de 6 mg./día produce síntomas extrapiramidales en un 30% de los pacientes, los cuales se van incrementando progresivamente con la elevación de las dosis hasta alcanzar un 50% con 16 mg./día, sugiriendo que el bloqueo de receptores 5-HT2 tiene una ventana protectora muy pequeña. Además, la Ritanserina, un antagonista de receptores 5-HT2 no logra antagonizar la catalepsia en ratas causada por el bloqueo de receptores D2 con Racloprida (Seeman, 1995).
Actualmente se considera que un antipsicótico atípico debe exhibir las siguientes propiedades: 1) disminuir los síntomas psicóticos, 2) disminuir los síntomas negativos, 3) disminuir los déficits neurocognitivos, 4) ser eficaz en pacientes refractarios, 5) pocos efectos extrapiramidales, 6) poca incidencia de discinesia tardía, 7) sin efectos en los niveles de prolactina
Estudios in vitro han demostrado que el predictor más importante de atipicidad de un antipsicótico es la disociación rápida de los receptores D2, teniendo mayor impacto que la alta afinidad a receptores 5-HT2. En condiciones basales 25% a 40% de los receptores D2 están ocupados por dopamina. Los bloqueadores D2 compiten por estos sitios de unión. Entre más rápida sea la disociación del fármaco y el receptor, más rápida es la respuesta del medicamento a las elevaciones de niveles de dopamina. Clozapina tiene una velocidad de disociación 100 veces mayor que haloperidol. Fuente:
Atípicos Depot:
Preparados depot de los atípicos en forma o no de estéres (esterificación de su grupo hidroxilo con un ácido graso de cadena larga), administrados por vía intramuscular, peligrosos por su lenta absorción y eliminación del organismo. Lo mismo dicho que en los típicos depot.
Benzamidas Sustituidas:
También llamados Ortopramidas, poco efecto extrapiramidal, efecto antiD2 a dosis muy elevadas, tienen mayor efecto de hiperprolactinemia: Por antagonismo dopaminérgico D2 a nivel tuberoinfundibular, se "libera" la inhibición dopaminérgica de la prolactina. Se presenta galactorrea, ginecomastia y amenorrea. Son enantiómeros de estos compuestos la metoclopramida (Primperan®, Reliveran®) y la cisaprida.
Derivados. 2-Pirrolidinil:
Análogos e Isómeros de Fenotiazinas
Son también compuestos tricíclicos, en los que se reemplaza el puente de S de las fenotiazinas por un puente de N = C y una cadena lateral cíclica. Tienen la ventaja de desarrollar muy escasos efectos extrapiramidales, mínimas reacciones distónicas y parkinsonianas, aunque pueden ocurrir otros efectos adversos: como fatiga, mareos, o hipertensión, taquicardia y convulsiones, que tienden a desaparecer lentamente con el uso. A veces se observa una sialorrea o salivación intensa. Menor acción antagonista dopaminérgica D2, bloqueante serotoninérgico 5-HT2A y dopaminérgico D4.
Dibenzodiazepinas:
De estructura similar a los antidepresivos tricíclicos
(pero el anillo central posee 7 miembros), con una sustitución piperazínica en el anillo central.
Dibenzotiazepinas:
Al igual que los derivados benzisotiazólicos, presentan
menor afinidad para el bloqueo de los receptores 5-HT2A que el resto de los antipsicóticos atípicos. Sin embargo, conservan la relativa menor afinidad para el bloqueo de los receptores D2.
Dibenzoxepinas:
Pierden el nitrógeno, de la estructura tricíclica o heterocíclica central. Mayor bloqueo de los receptores 5HT que los de la dopamina
Tienobenzodiazepinas:
Compuestos que se asemejan químicamente a la clozapina, compartiendo alta afinidad para el bloqueo de los receptores 5-HT2A y una menor afinidad por los receptores D2.
Benzisoxazol:
Contiene una estructura benzisoxazol + piperidina. Combinan alta afinidad por los receptores dopaminérgicos D2 y por los receptores serotonérgicos 5-HT2A en dosis bajas, no tienen casi efectos sobre los receptores muscarínicos
Benzisotiazol:
Presentando una estructura de benzisotiazol + piperazina. Alta afinidad para el bloqueo de los receptores 5-HT2A y moderada a baja afinidad para el bloqueo de los receptores D2, H1y alfa1 adrenérgicos.Indoles
Derivados Indólicos:
Tienen el núcleo indólico
La introducción de un metilo en el nitrógeno indólico supone un incremento notable de sus propiedades antiserotonínicas.Derivan su efecto inhibidor selectivo sobre las neuronas dopaminérgicas mesolímbicas y es debido a que ejerce efectos inhibidores equilibrados sobre los receptores centrales D2 de la dopamina y 5HT2 de la serotonina, así como sobre los receptores a1 adrenérgicos. Tienen mucha menos afinidad para los receptores D2 que la mayoría de los agentes antipsicóticos y tiene relativamente baja afinidad para los receptores D1. Tiene sólo de baja a moderada afinidad por los receptores colinérgicos y alfa-adrenérgicos e histamínicos H1
2- Imidazolidinona:
Alcaloides de la Rauwolfia:
Los alcaloides (de álcali y -oide) son compuestos orgánicos nitrogenados de carácter alcalino producidos casi exclusivamente por vegetales (aunque también los hay producidos por animales y hongos, y otros sintetizados químicamente). Normalmente derivan de los aminoácidos.
Agonistas Parciales:
En farmacología, los agonistas parciales son fármacos que se unen y activan un dado receptor, pero que tienen sólo una eficacia parcial en el receptor con respecto a un agonista completo. También es posible considerarlos ligandos que muestran tanto efectos agonistas como antagonistas: cuando un agonista completo y un agonista parcial están presentes al mismo tiempo, el agonista parcial actúa como un antagonista competitivo, compitiendo con el agonista completo por la ocupación del receptor y produciendo una disminución neta en la activación del receptor observado con el agonista completo solo. Clínicamente los agonistas parciales pueden utilizarse para activar receptores para producir una respuesta submáxima deseada cuando no hay cantidades endógenas adecuadas del ligando, o pueden reducir la sobreestimulación de los receptores cuando hay cantidades excesivas del ligando.
Quinolinonas:
Derivado de la estructura de la quinolinona. Agonismo parcial sobre los receptores D2 y 5-HT1A y antagonismo 5-HT2A
Azapironas:
Derivado de la estructura de la azapirona. Agonismo parcial sobre el receptor 5- HT1A y antagonismo 5 HT2A. Ver también ansiolíticos, azapironas
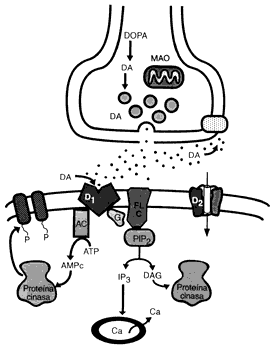
CICLO SINAPSIS DE LA DOPAMINA
Durante la actividad neuronal, la Dopamina es liberada de sus vesículas de almacenamiento. La cantidad almacenada y liberada depende de la capacidad disponible almacenada, de la proporción en que las vesículas son descargadas y recargadas y de la proporción en que nuevas vesículas son formadas.El incremento en la densidad de los receptores D2 siguiendo la administración crónica de antagonistas podría ser responsable del desarrollo de un desorden del movimiento denominado discinesia tardía.
Los agonistas a los receptores de dopamina, incluyendo las anfetaminas, bromocriptinas. Existe una fuerte correlación entre las dosis clínicas de los neurolépticos y su afinidad por los receptores D2 en el cerebro. Esto ha conducido a la hipótesis de que los desórdenes psicóticos son el resultado de una hiperestimulación de los receptores D2. La adminstración prolongada de neurolépticos a humanos o a animales de laboratorio pueden llevar a un incremento en la densidad de los receptores D2 del estriado y en la aparición de efectos secundarios extrapiramidales, incluyendo desórdenes del movimiento parkinsonianos y discinesia tardía. Las afinidades relativas de los receptores D2, D3 y D4 para los neurolépticos típicos y atípicos, conjuntamente con la expresión selectiva del ARNm para el receptor D3 en áreas límbicas del cerebro, ha conducido a la hipótesis de que la utilidad clínica de los neurolépticos en el tratamiento de enfermedades psiquiátricas puede ser debido, por lo menos en parte, a su capacidad para antagonizar la estimulación de los receptores D3 o D4, mientra que la disfunción motora observada al seguir un tratamiento crónico con neurolépticos típicos, podría ser debida a alteraciones en la densidad de los receptores D2 en el estriado.
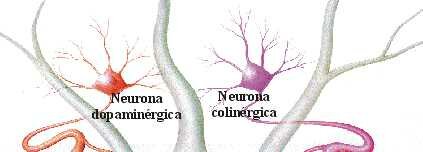
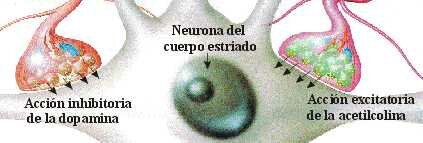
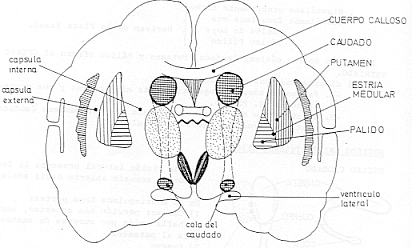
EQUILIBRIO DOPAMINA/ACETILCOLINA
Desde el núcleo caudado y el putamen, existe una vía hacia la sustancia negra que segrega el neurotransmisor inhibitorio GABA (ácido gamma aminobutírico). A su vez, una serie de fibras originada en la sustancia negra envía axones al caudado y al putamen, segregando un neurotransmisor inhibitorio en sus terminaciones, la dopamina. Esta vía mutua mantiene cierto grado de inhibición de las dos áreas y su lesión provoca una serie de síndromes neurológicos, entre los que se encuentra la enfermedad de Parkinson. Las fibras provenientes de la corteza cerebral segregan acetilcolina, neurotransmisor excitatorio, sobre el neoestriado. Las causas de las actividades motoras anormales que componen la enfermedad de Parkinson se relacionan con la pérdida de la secreción de dopamina por las terminaciones nerviosas de la sustancia negra sobre el neoestriado (tracto nigroestriatal) al que deja de inhibirlo. De esta forma, predominan las neuronas que segregan acetilcolina, emitiendo señales excitatorias a todos los núcleos de la base, responsables en conjunto, del planeamiento motor y algunas funciones cognitivas.
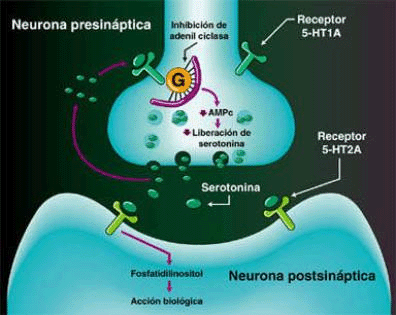
SEROTONINA
La clásica terapéutica antipsicótica se basa en el bloqueo dopaminérgico (D2) logrado con los neurolépticos tradicionales, con evidente acción sobre los síntomas positivos, pero escasa o nula sobre los negativos. A partir de la aparición de la Clozapina, primer antipsicótico atípico con acción predominantemente antagónica sobre los receptores 5HT2, se abre una nueva perspectiva en el tratamiento de la esquizofrenia. Se logra un efecto resocializante, antiagresivo, antiautístico, sin dejar de lado el efecto antidelirante y alucinolítico, y con escasa producción de efectos extrapiramidales. Este fármaco bloquea los receptores D2 mesolímbicos sólo en un 20%, mientras que lo hace en el 90% de los 5HT2.
En los cuadros psicóticos en los cuales predomina el componente disperceptivo, tienen su indicación específica los antagonistas 5HT3 (Ondansetrón, Granisetrón y otros). Añaden a su efecto alucinolítico una acción antidepresiva leve, y una acción neuroprotectora sobre la esfera afectiva y cognitiva.Receptores para la serotonina
No existe un receptor único para Serotonina, sino mas bien ha sido descrita toda una superfamilia de receptores con funciones específicas en las áreas pre y postsinápticas. Estudios farmacológicos y fisiológicos han contribuido a la definición de muchos subtipos de receptores para serotonina. Inicialmente se diferenciaron dos receptores diferentes de 5-HT en el íleon, llamados receptores D (bloqueado por dibencilina) y M (bloqueado por morfina). El receptor D se pensó que estaba en el músculo liso del íleon mientras que el receptor M, se consideró que estaba en la estructura ganglionar.
El desarrollo del ensayo de unión al radioligando fue propuesto por Pertoutka y Snyder en 1979 para etiquetar dos clases de receptores serotoninérgicos en el cerebro. Los lugares de unión con alta afinidad por [3H]-5-HT fueron designados como receptor 5-HT1; los lugares de unión etiquetados con alta afinidad por [3H]espiperona fueron denominados como receptor 5-HT2.
Las neuronas serotoninérgicas, originarias de los núcleos del rafe, ejercen un tono inhibidor sobre la transmisión dopaminérgica en las zonas nigroestriatales y mesocorticales limitando su síntesis y liberación. En la esquizofrenia esta inhibición de la dopamina por el control serotoninérgico está exagerada, lo que explica en parte la hipoactividad dopaminérgica nigroestriatal y mesocortical Esta inhibición puede ser levantada por las moléculas antagonistas serotoninérgicas.
La principal razón de la superioridad de los nuevos antipsicóticos con respecto a los neurolépticos convencionales es su propiedad antagonista 5-HT2 . El antagonismo de la actividad serotoninérgica en las zonas nigroestriatales y frontales permite una disminución de los síntomas deficitarios y una disminución de los efectos extrapiramidales, conduciendo a una mejor tolerancia.
Enlaces:
|
Dopamina
|
Serotonina
|
Noradrenalina o (Norepinefrina)
|
Histamina
|
 |
 |
 |
 |
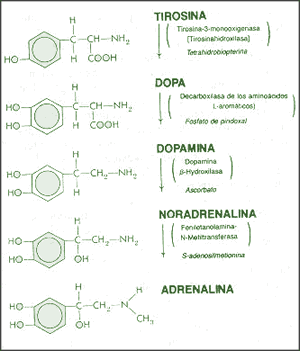

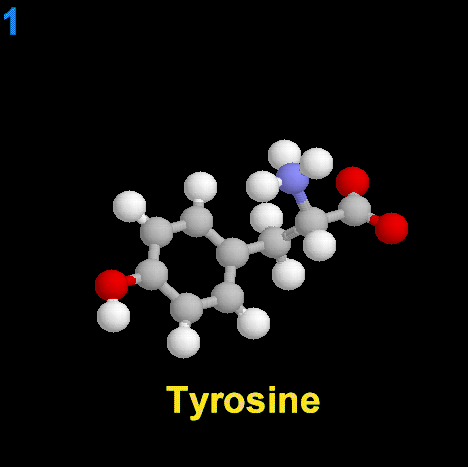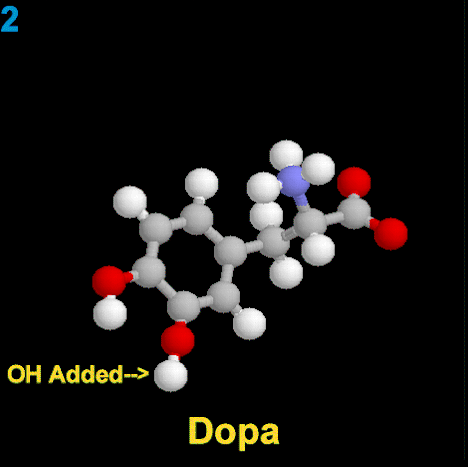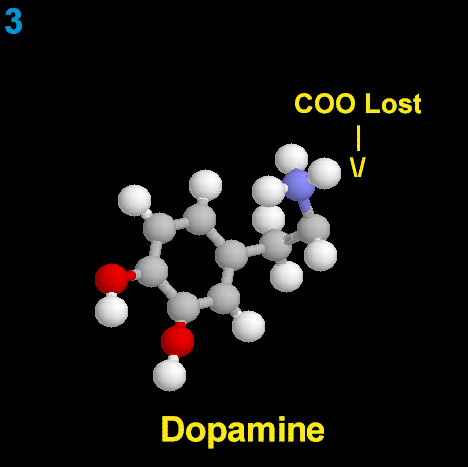
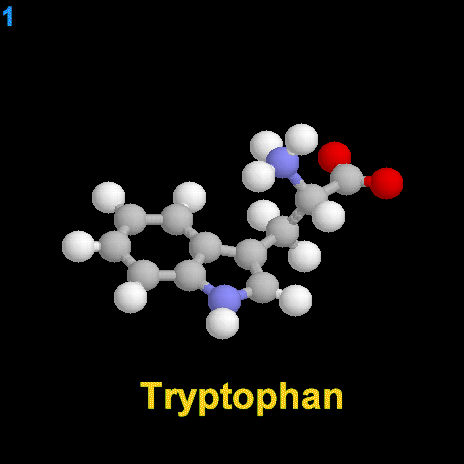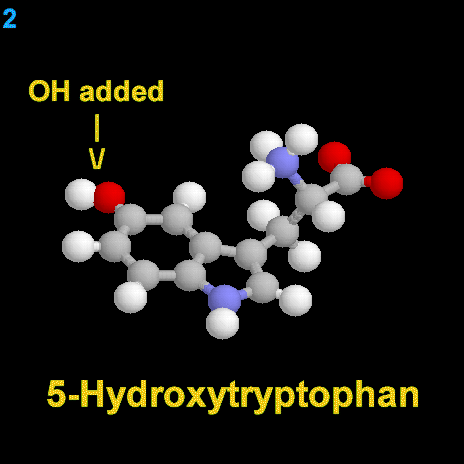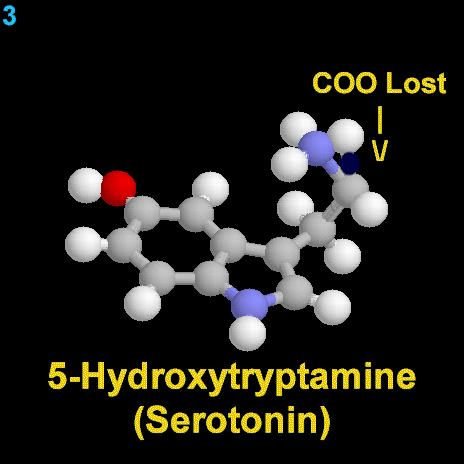
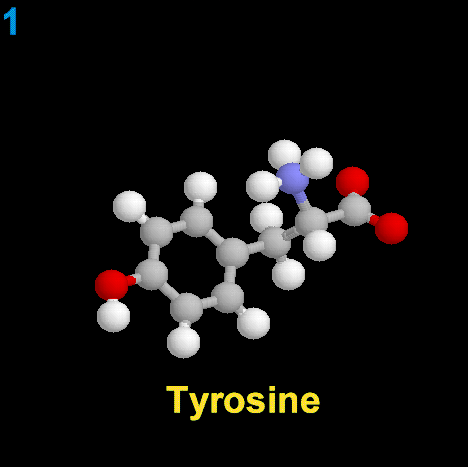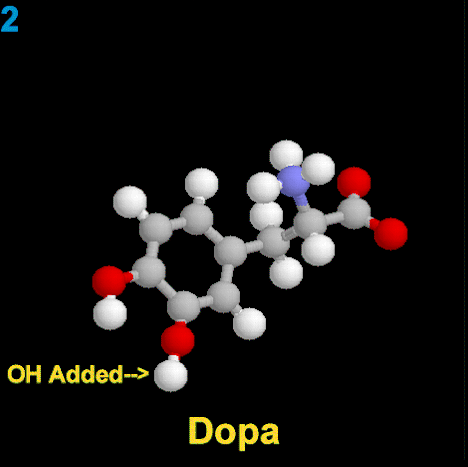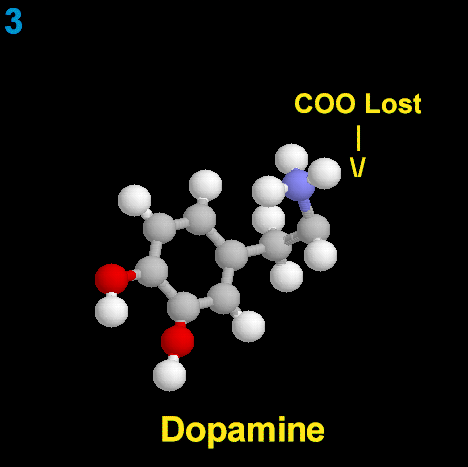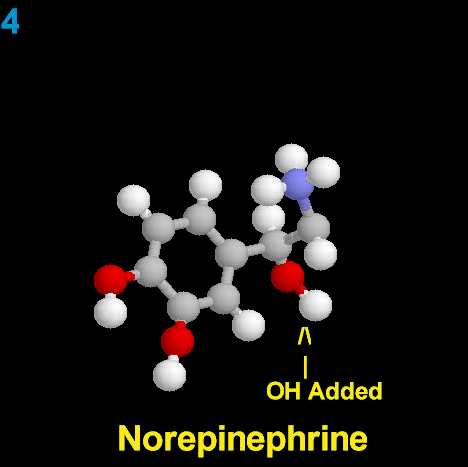
La dopamina es una monoamina del grupo de las catecolaminas, pues además del grupo amino solitario (-NH2), característico de las monoaminas, posee un grupo catecol, conformado por un anillo bencénico y 2 grupos hidroxilo (-OH).
La Dopamina es el precursor metabólico inmediato de la Noradrenalina y Adrenalina, sintetizada a partir de Tirosina que, por acción de la enzima Tirosinhidroxilasa es hidroxilada para convertirse en DOPA, que posteriormente es descaboxilada por la DOPA-descarboxilasa para dar Dopamina. Debido a su posición central en el metabolismo de las catecolaminas puede encontrarse Dopamina en cualquier lugar que se produzca Adrenalina o Noradrenalina. No obstante, su concentración a nivel del núcleo nigroestriado es mucho mayor que la de Noradrenalina. También hay neuronas dopaminérgicas en la retina, pero su función hasta la fecha es desconocida.
La biosíntesis de la dopamina está intrínsecamente relacionada con la NA, aunque su degradación está sujeta a los mismos sistemas enzimáticos, difiere en función de cual sea el primer sistema enzimático que actúe. Si actúa la monoaminooxidasa (MAO) se produce el ácido 3,4 dihidroxifenilacetico (DOPAC), que es un metabolito final de la dopamina; si actúa la COMT, hay un metabolito intermediario, la 3-metoxitiramina, momento en el cual interviene la MAO produciendo el producto final de la degradación, el ácido homovanílico (AHV).
La liberación es similar a la de la NA, siendo las anfetaminas unos potentes liberadores. Son inhibidores los a-hidroxibutiratos y la reserpina, que es también un depleccionante de la DA al igual que ocurría con la NA.
Por otra parte, la recaptura no debe seguir el mismo camino que la NA, puesto que los antidepresivos tricíclicos no la afectan y sin embargo, sí la anfetamina y la benzotropina.
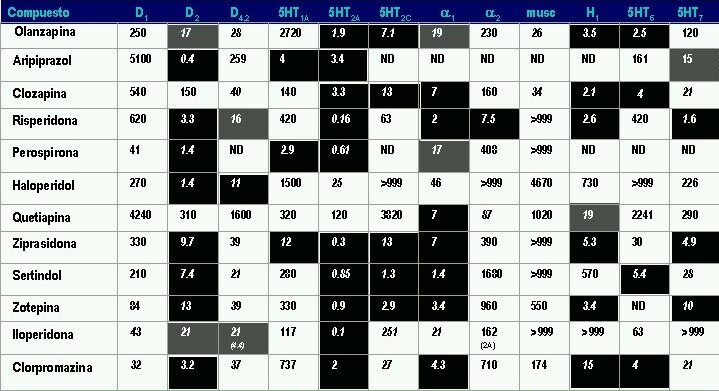
Las afinidades de los APs por diferentes receptores según su constante de inhibición (Ki) expresada en nanomoles se presenta en la siguiente tabla. En fondo negro se aprecian los receptores sobre los que cada uno de estos medicamentos presenta mayor afinidad (menor Ki) y en gris los que presentan moderada afinidad
Afinidad Receptores
Según constante de inhibición (Ki) expresada en nanomoles
Para comparar de forma gráfica la afinidad de diferentes antipsicóticos ir a --> búsqueda taxonómica de esta web.
|
Nombre:
|
Gráfico y Leyenda: |
|
Dopamina D1
|
Dopamina D5
|
Dopamina D2
|
Dopamina D3
|
Dopamina D4
|
|
|
Serotonina 5-HT1A
|
Serotonina 5-HT2A
|
Serotonina 5-HT2B
|
Serotonina 5-HT2C
|
Serotonina 5-HT6
|
Serotonina 5-HT7
|
|
Noradrenalina a1
|
Noradrenalina a2
|
*Acetilcolina M1-5
|
Histamina H1
|
||
Cuanto < más bajo es el valor, más > afinidad del receptor hay. Los valores difieren según la fuente farmacológica aquí son mayormente de Jesús Flórez completados con la tabla de arriba.
Antagonismo D2 = efecto sobre síntomas positivos [todos los antipsicóticos]; Agonismo 5-HT1A: efecto deletéreo en cognición [Aripiprazol]; Antagonismo 5HT2A: efecto favorable en cognición por normalización del funcionamiento NMDA [todos los atípicos y clorpormazina]; Antagonismo 5-HT2C: aumento del apetito [clozapina, olanzapina]; Antagonismo alfa 1: hipotensión, efecto sedante y mejoría de memoria de trabajo bajo estados de estrés [todos los antipsicóticos atípicos]; Antagonismo alfa 2: hipertensión [risperidona]; aumenta la liberación de noradrenalina; Efecto anticolinérgico central: compromiso de funciones cognoscitivas [clozapina]; Antagonismo H1: sedación, aumento de apetito [atípicos y clorpromazina]; Antagonismo 5-HT6: elevación de acetilcolina en espacio sináptico en corteza prefrontal e hipocampo (mejoría cognoscitiva)[olanzapina y clozapina] . En los receptores de acetilcolina, la mayor parte de fuentes hacen una media de los 5 receptores acetilcolinérgicos del tipo M1 (M1,M4,M5) activadores de la fosfolipasa C (Gq) y los del tipo M2 (M2,M4) inhibidores de la adenilato ciclasa (Gi), acciones contrapuestas pero dan el dato medio.
Fuente: psicofarmacologia.info (modificado)
Ver gráfico afinidad a receptores de algunos antipsicóticos atípicos
Enlaces:
Velocidad de disociación de los antipsicóticos
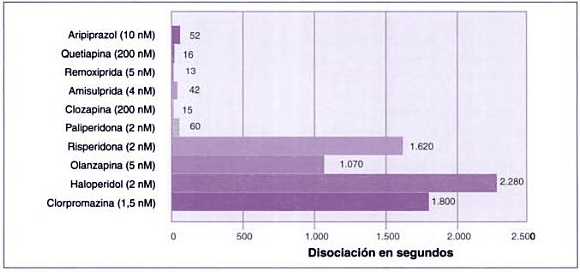
Velocidad de disociación de diversos antipsicóticos del receptor D2 (50%). Modificada de Seeman (2005).
Fuente del gráfico: Libro "Las esquizofrenias: sus hechos y valores clínicos y terapéuticos" 2007 de Alfonso Chinchilla Moreno.
Imagen: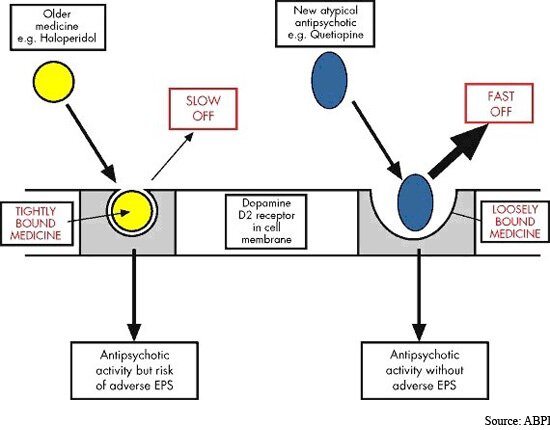
Todos los fármacos antipsicóticos disponibles actualmente se unen a los receptores de dopamina. Los antipsicóticos típicos se unen fuertemente y disocian lentamente del receptor de la dopamina, mientras que los fármacos atípicos se unen más libremente y disocian más rápidamente.
|
grupo D1
|
grupo D2
|
|
D1 |
D5 |
D2 |
D3 |
D4 |
|
|
Agonistas |
skf 38393 |
Dopamina |
Bromocriptina |
Dopamina |
Dopamina |
|
Antagonistas |
Alfa-Flupentixol |
Haloperidol |
Sulpiride |
Clozapina |
|
|
Distribución |
N. Estriado |
Hipotálamo |
N. Estriado |
T.olfatorio |
CORTEX FRONTAL |
 Transducción de la señal
Transducción de la señal
Ejemplo de receptores unidos a proteínas G estimulando a la Adenilato Ciclasa (Gs)
Hay otro tipo de transducción con proteínas G que estimula a la fosfolipasa C (PLC) (G0), con la aparición de IP3 y DAG como segundos mensajeros: verlo en estimulantes apartado varios.

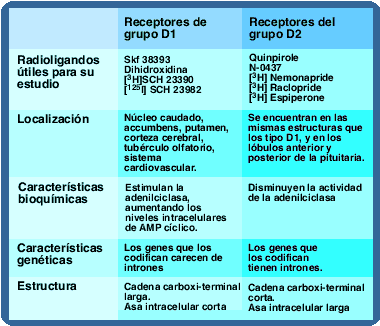
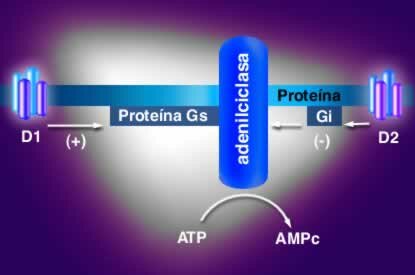
Acción de los receptores tipo D1 y D2 sobre la adenilciclasa.
Todos los receptores dopaminérgicos son ligados a proteína G, la diferencia entre los dos grupos radica en el tipo de proteína G y los agentes mensajeros utilizados. Todos los receptores ligados a proteína G constan de una porción extracelular que tiene el grupo amino-terminal, una porción proteínica que atraviesa la membrana 7 veces y una porción intracelular que posee la terminación carboxilo que con frecuencia e asocia a un grupo palmitol.
Existen diferencias en la longitud de las asas intracelulares y las terminaciones carboxílicas entre los receptores tipo D1 y los receptores tipo D2, en los primeros las asas intracelulares son cortas, y las terminaciones carboxílicas largas, mientras los D2 tienen asas intracelulares largas y terminaciones carboxílicas cortas. Adicionalmente, existen diferencias entre los genes que los codifican los de receptores D1 carecen de intrones. También estimulan la síntesis de AMP cíclico mediada por proteínas asociadas a nucleótidos de guanina llamadas proteínas G, del subtipo estimulatorio (Gs).
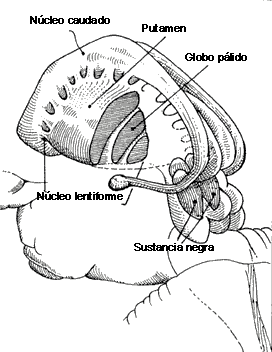
Ambos tipos de receptor son muy comunes en el núcleo caudado, putamen y núcleo accumbens. Los receptores D1 se encuentran además en neuronas postsinápticas del estriado, la región medial del globo pálido y en la parte reticulada de la sustancia nigra.
La mayoría de receptores D2 en el estriado actúan como autorreceptores. Estudios post mortem de pacientes esquizofrénicos han mostrado aumento en la densidad de receptores D2 a este nivel, lo que probablemente se debe al tratamiento crónico con antipsicóticos pues los estudios imagenológicos in vivo de pacientes que no han recibido tratamiento farmacológico nunca, no fueron consistentes con este hallazgo.
En el hipocampo, los receptores D1 inducen la producción tardía de proteínas, responsable de los mecanismos de potenciación del estímulo.
Los receptores D2 están asociados a proteínas G del subtipo inhibitorio (Gi).
La activación de los receptores tipo D1 produce activación de adenilciclasa, aumentando las concentraciones de AMP cíclico , responsable del aumento marcado de la concentración de calcio intracelular, que al parecer depende de liberación del elemento de las reservas intracelulares. Este fenómeno es importante en la liberación de dopamina al espacio sináptico.
Por otra parte, la activación de los receptores tipo D2, inhibe adenilciclasa, estimula a mitogénesis y produce acidificación del medio extracelular. Estos receptores D2 son más comunes en el cerebro humano que los D1.
La mayoría de los fármacos antipsicóticos tiene mayor afinidad por uno de los 2 grupos. Las tioxantinas y fenotiazinas tienen mayor afinidad por los receptores D1, mientras las benzamidas y butirofenonas prefieren los receptores D2. La clozapina muestra una moderada selectividad sobre los receptores denominados antiguamente D4, que pertenecen al grupo tipo D2.
![]() Enlaces:
Enlaces:
- ,
Funcionamiento de los Núcleos de los Ganglios de la Base y Sustrato neural
Hay un dibujo en la sección varios 2 apartado COREAS que de un vistazo se capta la explicación de cómo funciona el modelo de los antipsicóticos bloqueo D2 del Globo pálido externo de la vía indirecta
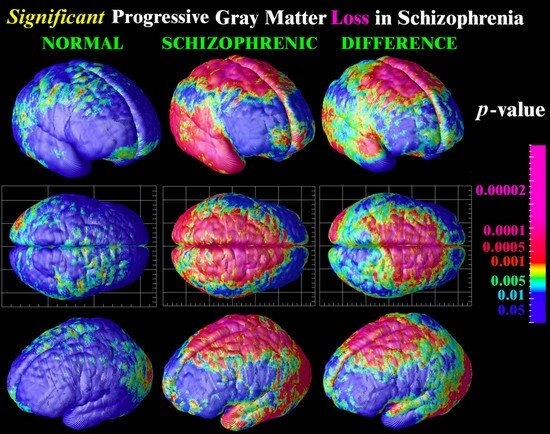
La pérdida de tejido cerebral en un intervalo de cinco años, la pérdida grave se muestra en color rosa. Los pacientes con esquizofrenia (derecha), más rápido pierden tejido cerebral que sus compañeros sanos (izquierda).
Imagen cortesía de Paul Thompson, Laboratorio de Neuro Imaging, la Universidad de California.
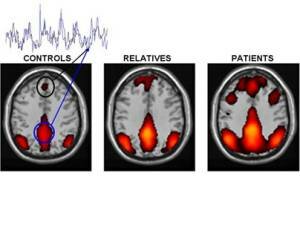
Alteración de la conectividad del cerebro en personas con esquizofrenia y familiares de primer grado. Zonas de color representan una red interconectada de regiones del cerebro que muestran una actividad sincronizada (superposición de huellas en color negro y azul) cuando los sujetos descansan y permiten que su mente vague. El importe de la sincronía, que refleja la fuerza de las conexiones funcionales entre los distintos ámbitos, está aumentada en pacientes con esquizofrenia. Los parientes en primer grado de las personas con la enfermedad también muestran un cierto aumento, aunque menos que los pacientes. Círculo Negro: la corteza prefrontal medial. Círculo Azul corteza cingulada posterior / precuneous. Crédito de la imagen: Susan Whitfield-Gabrieli, McGovern Institute for Brain Research del MIT
Los voluntarios fueron escaneados mediante resonancia magnética funcional por imágenes (fMRI), mientras descansaban y mientras ejecutaban tareas de memorización fáciles o difíciles. El punto de vista tradicional sobre la esquizofrenia es que los pensamientos, percepciones y emociones alteradas que caracterizan la enfermedad están causados por desconexiones entre las regiones cerebrales que controlan estas diferentes funciones.
Pero este estudio ha desvelado que la esquizofrenia también involucra un exceso de conectividad entre las regiones cerebrales involucradas en la reflexión sobre uno mismo y que devienen activas cuando no estamos pensando sobre nada en particular, o cuando pensamos sobre nosotros mismos.
Normalmente, las personas suprimen este sistema cuando realizan tareas que demandan mucha atención, pero Susan Whitfield-Gabrieli (MIT), John D. Gabrieli (MIT), Larry J. Seidman (Escuela Médica de la Universidad de Harvard), y los otros 10 autores del estudio, encontraron que los pacientes con esquizofrenia no actúan de esta manera.
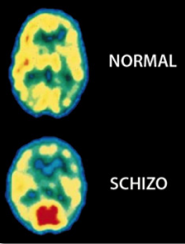
Tomografía por Emisión de Positrones (TEP) del cerebro muestran grandes diferencias en la distribución de la actividad eléctrica en un cerebro normal, comparado con el cerebro de un paciente esquizofrénico (derecha). La esquizofrenia afecta a aproximadamente 1% de la población mundial. Reproducido con permiso de. Photo Researchers, Inc.
Todas las diferencias Neuroanatómicas y funcionales de la Esquizofrenia
ESQUIZOFRENIA ENLACES:
Afinidad al receptor D2 de los antipsicóticos:
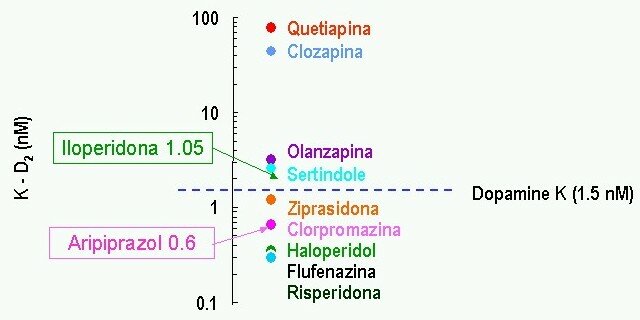
Lawler C, et al. Neuropsychopharmacology 1999; 20:612-27; Corbett R, et al. CNS Drug Reviews 1997; 3:120-47 ![]() El mismo gráfico pero más completo
El mismo gráfico pero más completo
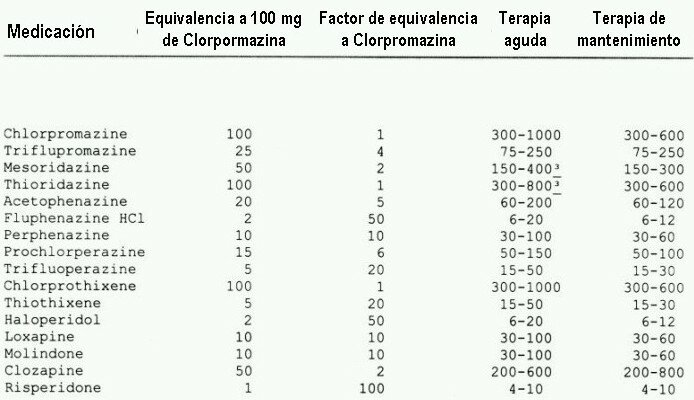
(adaptado de Zito 1994 & Kane 1996)
Gráficos sacados de

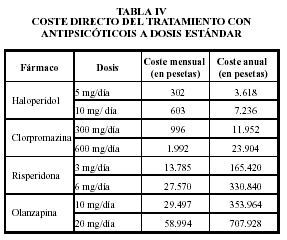
Gráficos de
Perfil Antipsicótico
|
Nombre
|
Potencia antipsicótica
|
Sedación
|
Efectos vegetativos
|
Síntomas extrapiramidales
|
|
Levomepromazina
|
++
|
+++
|
+++
|
+
|
Leyenda: (+) muy leve, + leve, +(+) moderado, ++ intenso, ++(+) potente, +++ muy potente
* Fuente de la búsqueda: farmacología humana, Jesús Flórez
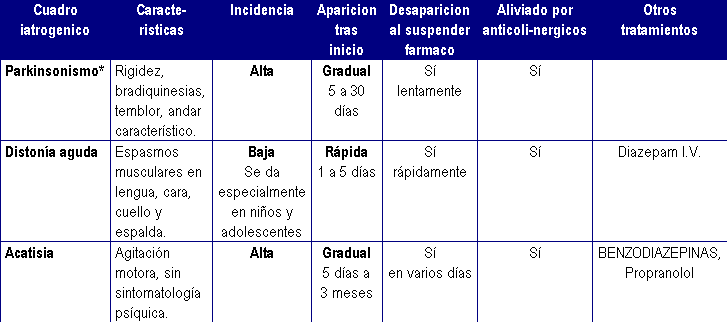
Principales reacciones extrapiramidales por antipsicóticos
Fuente:
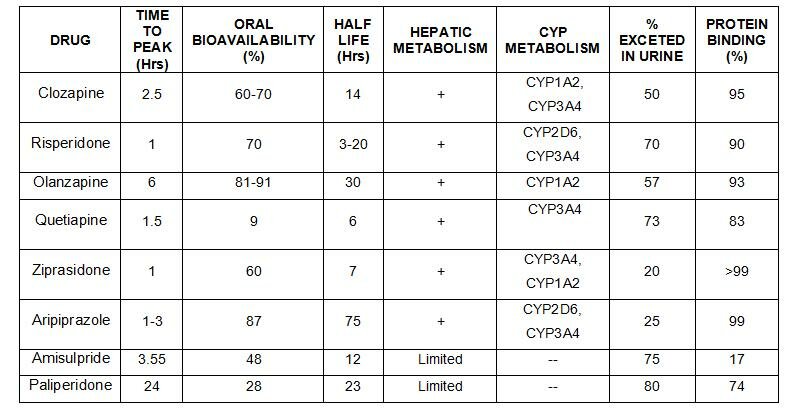

- Absorción
- Vía oral: pico máximo plasmático, 2-4 horas (absorción modificada por café, té, anticolinérgicos, antiácidos).
- Vía intramuscular: pico máximo, 20-30 min.
- Se desaconseja la vía intravenosa.
- Distribución
- Gran variabilidad interindividual de niveles plasmáticos (entre 10-100 veces; efecto del primer paso hepático).
- Unión a proteínas plasmáticas en un 90-98%.
- Alta lipofilia. Alta biodisponibilidad. Afinidad preferente en SNC, pulmones, tejidos altamente vascularizados.
- Eliminación
- Metabolización preferente en microsomas hepáticos:
- Glucuronoconjugación
- Hidroxilación
- Sulfóxidos
- Eliminación renal:
- 1/2 a (Vida media de distribución): 2 horas (oral)
- 1/2 ß (Vida media de eliminación): mayor de 30 horas (oral)
Su absorción tras la administración oral es rápida (2-4 horas) pero susceptible a alteraciones por diferentes factores como son los antiácidos, los anticolinérgicos, el café y el té, que actúan retardando su absorción. Se recomienda el intervalo de 2 a 4 horas entre el consumo de estos productos y la administración de un antipsicótico. La absorción por vía intramuscular es todavía más rápida (10-30 minutos). Si se precisa un efecto inmediato, se recomienda el músculo deltoides por su irrigación 3 veces superior a la musculatura glútea. Debido a su elevada fijación a las proteínas plasmáticas (90-98% del total), su molécula activa, es decir la fracción libre, aumenta considerablemente en situación de hipoproteinemia.
Se metabolizan en el hígado, basicamente en el sistema citocromo P-450. Su eliminación es urinaria y en menor proporción biliar. Con la edad el tiempo de eliminación aumenta, produciendo un mayor riesgo de acumulación del fármaco.
Alcanzan niveles plasmáticos estables en 5-10 días. Su vida media oscila entre 10 y 24 horas, facilitando así una cómoda administración de una sola dosis diaria cuando el paciente logra una condición estable.
Todavía persiste la controversia sobre la relación entre niveles plasmáticos de los antipsicóticos y su respuesta clínica. Los resultados hasta ahora obtenidos al respecto no son concluyentes. Tampoco parece definitiva la existencia de "ventanas terapéuticas" para los antipsicóticos.
3 hipótesis; hipoactividad, hiperactividad y neurotoxicidad mediada por glutamato
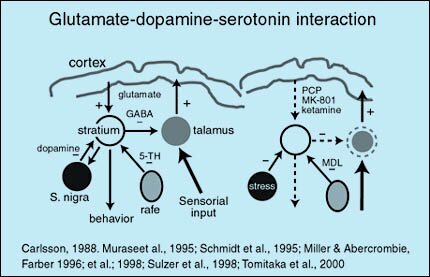
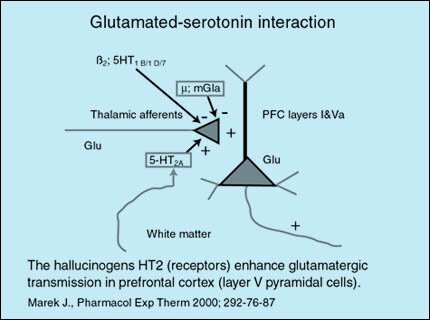
Rol del glutamato
El glutamato es el neurotransmisor excitatorio más importante del sistema nervioso central, estimulando todas las neuronas de una forma no específica.
Es sintetizado a partir de glutamina por la enzima mitocondrial, glutaminasa y luego almacenada en vesículas hasta su liberación.
El glutamato liberado por las neuronas puede tomar 2 vías: a) una parte es recaptada en la membrana presináptica mediante mecanismos altamente específicos, b) gran parte es captada por los astrocitos, la mayoría del glutamato glial es convertido en glutamina, que es transportada a la hendidura sináptica donde es convertido nuevamente a glutamato.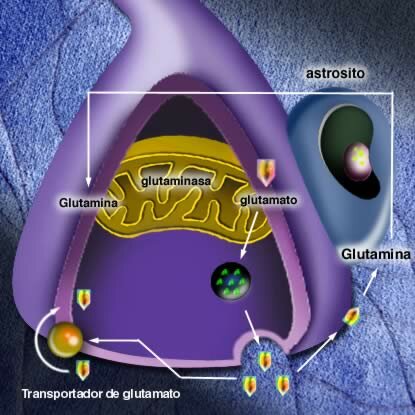
La liberación de glutamato en la hendidura sináptica es inhibida por drogas anticonvulsivantes como lamotigrina.
Hay 6 tipos de receptores postsinápticos de glutamato, 3 de ellos están unidos a canales iónicos, y los otros 3 son asociados a proteína G.
Los receptores asociados a canales iónicos son el N-metil-D-aspartato (NMDA), el a-amino 3-hidroxi 5-metilisoxazol 4-ácido propiónico (AMPA) y el receptor de Kainato. Los receptores asociados a proteína G son los receptores metabotrópicos tipo I, II y III.
El NMDA está ligado a un canal de calcio dependiente de voltaje. En estado de reposo no es activado por glutamato, pues un ion magnesio lo bloquea, cuando la membrana se despolariza, el magnesio se libera, permitiendo la entrada de calcio.
 Este tipo de receptor se encuentra en todo el encéfalo, pero en mayores cantidades en el hipocampo y el neocórtex. Se ha asociado con procesos de aprendizaje y muerte neuronal cuando permanece despolarizado por períodos prolongados.
Este tipo de receptor se encuentra en todo el encéfalo, pero en mayores cantidades en el hipocampo y el neocórtex. Se ha asociado con procesos de aprendizaje y muerte neuronal cuando permanece despolarizado por períodos prolongados.
Anestésicos disociativos como la ketamina, y sustancias estimulantes como la fenilciclidina actúan a este nivel.
Se cree que los receptores NMDA tiene un rol importante en la patogénesis de la esquizofrenia, basados en la observación de los síntomas psicóticos producidos en casos de intoxicación por fenilciclidina. También se ha postulado que las alteraciones de la esquizofrenia son causadas por muerte neuronal acelerada mediada por estos receptores.
Los receptores de kainato se encuentran en cantidades abundantes en el hipocampo, están asociados a un canal de sodio que al permanecer abierto por tiempos prolongados, permite una entrada masiva del catión produciendo muerte neuronal por sobrecarga osmótica. En estudios de pacientes con esquizofrenia se han encontrado mayores concentraciones de este tipo de receptor en la corteza frontal, y disminución de los mismos en el hemisferio izquierdo.
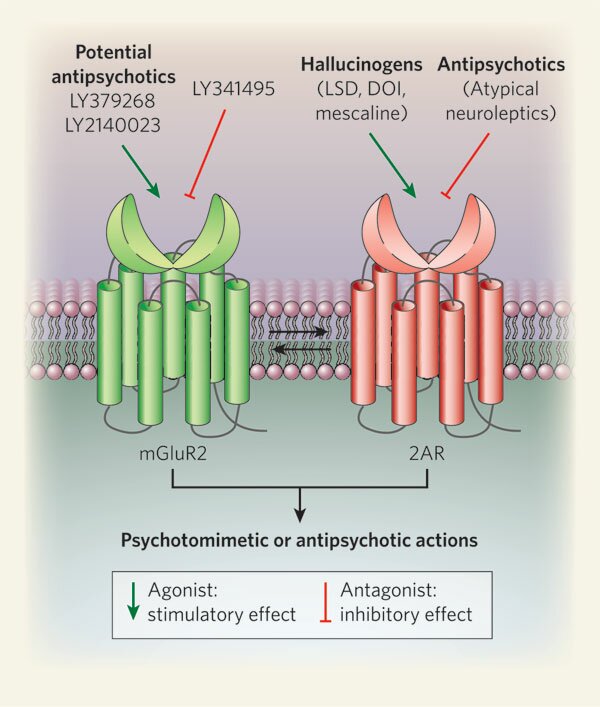
Nuevos Antipsicóticos agonistas receptor mGluR2 antagonizan 2AR o receptor de la serotonia 5-HT2
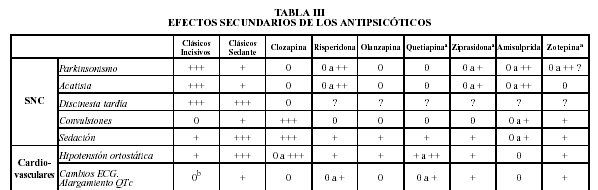
EFECTOS ADVERSOS DE LOS ANTIPSICOTICOS
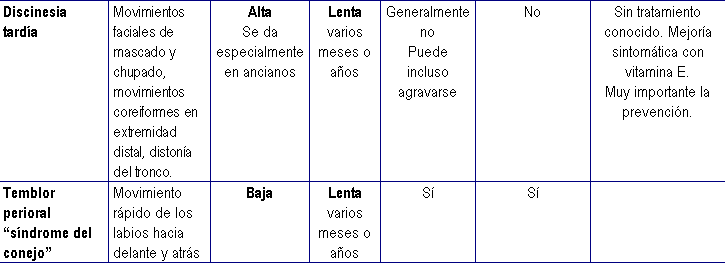
NEUROLOGICOS----------- Distonía aguda. Acatisia. Parkinsonismo. Temblores periorales (Sdr. "del conejo"). Acinesia. Discinesia tardía. Disnea e Hiperventilación
GASTROENTEROLOGICOS--- Sequedad de boca. Sialorrea. Transtornos de la motilidad esofágica. Constipación. Ileo paralítico.
UROLOGICOS---- Retención urinaria. Tenesmo. Disuria. Polaquiuria.
HEPATICOS------ Colestasis intrahepática (ictericia). Hepatotoxicidad (clorpromacina).
CARDIOVASCULARES--------- Taquicardia. Hipotensión. Cambios ECG. Arritmias (tioridacina). * Algunas drogas antipsicóticas son bloqueantes de canales de calcio a nivel neuronal, del músculo cardíaco y del músculo liso. Por ejemplo, la tioridazina y la pimozida, lo cual puede explicar su mayor toxicidad cardíaca (prolongación del intervalo QT y taquicardia ventricular con riesgo de torsión de punta). Miocarditis en la clozapina por hipereosinofilia
HEMATOLOGICOS--- Leucocitosis. Eosinofilia. Aplasia medular. Trombocitopenia. Agranulocitosis o Neutropenia (clozapina) es la disminución aguda o crónica de granulocitos de la sangre, condición anormal de la sangre que puede predisponer al cuerpo humano a contraer infecciones.
ENDOCRINOS--------- Hiperprolactinemia. Amenorrea. Ginecomastia. Galactorrea (sulpiride). Aumento de peso.
Web interesante de los (estimuladores del apetito)
SEXUALES------- Transtornos de la eyaculación y/o erección. Pérdida de la libido. Frigidez.
DERMATOLOGICOS------- Dermatitis alérgica. Dermatosis por contacto. Fotosensibilidad. Urticaria. Decoloración de la piel. Pigmentación. Exantemas maculopapulares.
OCULARES------- Visión borrosa. Queratopatías. Cataratas estrelladas y planas (clorpromacina). Retinopatía pigmentaria (tioridacina). Empeoramiento del glaucoma.
S.N.C---------- Astenia. Sedación. Somnolencia. Disminución del umbral convulsivo. Delirium. Hipotermia (edad avanzada).
OTROS------- Síndrome Neuroléptico Maligno. Neumonía
Diabetes y sobrepeso
Diabetes tipo 2 y un gran aumento de peso, con causa multifactorial por bloqueo de diversos receptores a nivel central como periférico,
Una de las causas es que aumentan significativamente la actividad de la enzima AMP quinasa (AMPK) en el hipotálamo, región del cerebro que, entre otras funciones, regula el apetito por bloqueo de los receptores de la histamina H1. Estos dos elementos bioquímicos -el receptor de la histamina H1 y la enzima AMP quinasa- están relacionados con la regulación de la ingesta de comida.
Toxicidad Neuroléptica
Apoptosis neuronal
1. Toxicidad de los metabolitos de los antipsicóticos más en los típicos que en los atípicos, en las mitocondrias celulares produciendo muerte neuronal, concentración de monoaminas en el citoplasma e inhibición de la respiración celular mitocondrial por una selectiva inhibición de la NADH: ubiquinona oxirreductasa (en el complejo I).
2. Disminución de sustancia gris en el cerebro, retraso cognitivo, sensorial (retraso mental, retraso sensorial, además del motor) por disminución de la señal dopaminérgica Bloqueo D2 produce down regulation D1 en todo el cerebro e inhibiendo la producción de neutrofinas (plasticidad y desarrollo neuronal impidiendo apoptosis).
Por bloqueo receptores 5HT-2 y a1.
La familia de receptores 5HT2 está conformada por 3 subtipos de receptores ligados a proteína Gq, también el receptor a1, que estimulan el metabolismo de Un fosfoinosítido, inositol fosfato o simplemente inosítido, es un fosfolípido que contiene en su estructura uno o más inositoles modificados por adición de uno o más grupos fosfato. Poseen especial relevancia en biología celular puesto que actúan como segundos mensajeros en la transducción de señal de las células.
Verlo con más detalle: en antidepresivos y en estimulantes
Ver más efectos iatrogénicos: Prolongación del intervalo QT, Síndrome Neuroléptico Maligno, Aumento de Prolactina, Acatisia y disnea Respiratoria y otros.
![]() Enlaces:
Enlaces:
- (en alemán)
COMENTARIOS DE: Levomepromazina

Tengo 64 anos en diversas consultas con antidepresivos por problemas de depresion adicciones y el mas fuerte hasta ahora transtornos severos del sueno me iba relativamente bien con el sinogan pero los sintomas adversos como rigides en un brazo y falta de animos y ganas de salir entre otros,cambio de medico conllevo al cambio de medicamento uno por otro y por otro ahora no puedo dormir sino 5 despues cuatro horas y media quisiera poder dormir mas que me puede recomendar
Hola. Hace 6 meses que tomo Nozinan (levomepromazina) 25mg. Al principio tomaba 2 pastillas por dia. O sea 50mg diarios. Pero dormia muchas horas y con mi psiquiatra decidimos bajar la dosis a 1 pastilla por dia. El problema es que empecé a tener fotosensibilidad. La luz me da dolor de cabeza, especialmente las luces que parpadean. Y el dolor de cabeza me dura dias enteros, a veces 3 o 4 dias. Tambien ver luz (electrica o solar) me produce nauseas. Puede que este medicamento tenga algo que ver con esto de la fotosensibilidad?

El fármaco sí puede producir fotosensibilidad, que es una reacción alérgica de la piel a la luz del sol. Lo que usted se refiere del dolor de cabeza es Fotofobia, y si le pasa esto después de tomar el fármaco seguramente que será la causa
Al tratarse por lo general de una consecuencia o un desencadenante, lo más inmediato para remediar la fotofobia es terminar primero con lo que la ha originado. Así, si se trata de un efecto secundario de un medicamento, no habrá más que dejar de tomarlo o sustituirlo y así cesarán las molestias. Se intuye entonces, que dependiendo cuál haya sido la causa, el tratamiento será distinto. Por eso es esencial acudir al médico especialista y que sea él quien dictamine los pasos a seguir.
TENGO PROBLEMAS DE INSOMNIO, HE TOMADO DIFERENTES MEDICAMENTOS TANTO NATURALES COMO DE LA MEDICINA COMUN, PERO NO OBTENGO RESULTADOS, AHORA ME FORMULARON LEVOMEPROMAZINA, PERO TENGO MUCHO MIEDO DE HACERLO, PODRIAN ORIENTARME AL RESPECTO.
GRACIAS

México-Querétaro:
Me han recetado Levomepromazina de 1/4,1/2,3/4,1 (por un mes)..siguientes meses por un año 1.Al siguiente día del inicio me sentí con dolores de cabeza intensos, y mucho sueño siendo las 11:00 a.m.Adicionalemente tomo la misma dósis con Sertralina y por último 1/2 clonazepan..PERO YA NO QUIERO SEGUIR TOMANDOLOS, CADA VEZ ES MÁS DIFICIL SALIR Y NO HAGO NADA DE EJERCICIO porque no me dan desesos de hacerlo..TENGO 69 AÑOS Y EL IMSS (gobierno) MÉXICO ES PÉSIMO.DÁ CONSULTAS CADA AÑO..Y LA GENTE SE MUERE Ó SE VUELVE LOCA..¿¿QUE OPCIONES PUEDEN AYUDARME A DEJARLOS??..ME DIERON DURANTE 2 AÑOS VENAFLAXINA (2013-2014), ALPROZALAM, CLONAZEPAN EN DIFERENTES DOSIS YA NO CONTROLADAS..AHORA TOMO LO ANTERIOR..¡¡QUE ESPERO¡¡..EL SUICIDIO, LOCURA Ó QUE..SALUDOS....

{kind=link}
{kind=link}
{kind=link}
Hace cincuenta años que tomo sinogan 12mg. duermo muy bien y durante todo el dìa la paso muy bien. Actualmente tengo 72 años y me preocupan los efectos secundarios que pueda sufrir ahora. Estarè agradecido a quien me indique si debo continuar con el fàrmaco o reemplazarlo.
he sufrido de depresiones por eso tomo remedios psiquiátricos, entre ellos NOZINAN supuestamente para dormir, pero duermo escasas 5 horas. mi edad 61. gracias....
pues a mi me lo recetaron la cosas es que yo tengo hepatitis c y pues es solo por un tiempo despues de 5 o 6 años como que recaigo pero en ese tiempo estoy fuerte y de lo mejor ya que a la mejor me dan psicoterapia, y a mi me lo recetaron por un trastorno de panico, ok saludos y a ver que pasa
Si es verdad que el sinogan tiene esos inconvenientes; yo aconsejaria probar con perfenazina, (Decentan), a ver que tal. saludos
ME ME MEDICZRON OLANZAPINA Y ME DIO MUCHISISMO USUEÑO PORQUE YA ESTANDO EN TERAPIA DE DOBLE AA YA NO LA CONSU'I DL PSIQUITRA ME DUO UNA AEQUIVOCADA MEDICACION QUE ESTO ME LLEVARIA A LA LOCURA J HOTA ME SIENTO MAS TRANQUILO YA QUE ESTO ES PARA QUE TENGAN EN CUENTA QUE TIENEN QIE HACER UN ESTUDIO NEUROLOGICO MAS PROFUDO EL TEST A VECES NO RESULTA Y EL ESSTUDIO ENCEFALOGRAMICO ASI COMO SE MUESTRA SANGRE PARA LABORATORIO ASI EA CON UN PACIENTE PSISQIATRICO
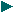 |
 |
Comentarios 1 a 10 de 14
6 meses para responder posts antiguos
¡Queremos tu opinión!
Si pregunta sobre este fármaco revise antes estos dos enlaces en particular
Ahora ya puede entender el prospecto y la ficha técnica