Búsqueda Simple
Búsqueda Completa
Taxonómica
Anatomía

+ Teoría
Patología
Aleatorio Principio Activo/ComercialFármacos o Drogas Más Visitadas
Levomepromazina

707355 visitas
Desvenlafaxina

273480 visitas
Clobenzorex
271940 visitas
Quetiapina

239062 visitas
Acepromazina
208539 visitas
Últimos comentarios

Sigue las noticias por

Total Fármacos Visitados
15.037.655
Desde Noviembre de 2008
CLASIFICACIÓN DE ANSIOLÍTICOS/SEDANTES/HIPNÓTICOS


Nombre: Adinazolam
Comercial: Deracyn
Foto:
Fórmula: 
Fármaco consultado 9332 veces
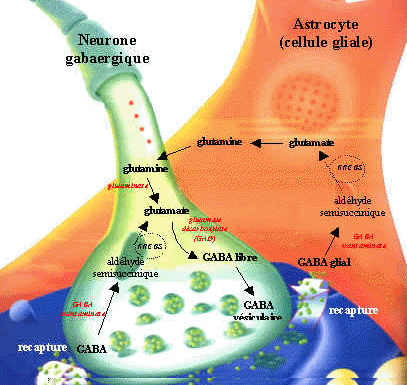
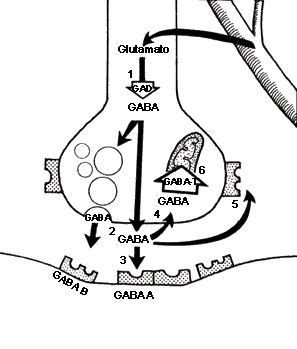
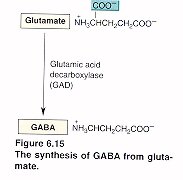
Síntesis y degradación de GABA
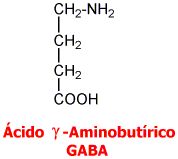
Ácido Gamma Aminobutírico GABA:
El GABA externo no pasa la barrera hematoencefálica.
El GABA es sintetizado a partir de la descarboxilación del Glutamato, mediada por la enzima Glutamato Descarboxilasa (GAD) que tiene como coenzima el piridoxal fosfato (forma activa de la vitamina B6). Una vez sintetizado, el GABA es introducido en vesículas y está listo para salir de la neurona presináptica. Cuando se produce el estímulo nervioso, GABA es liberado de la neurona presináptica y llega hasta la neurona postsináptica donde es reconocido por los receptores GABAA y GABAB. El GABA que no interacciona con los receptores es recaptado bien sea por la célula presináptica o por las células gliales. Una vez allí, mediante la GABA Transaminasa es degradado el GABA a ácido succínico semialdehído que la Succínico semialdehído deshidrogenasa (SSADH) lo convierte a ácido succínico. La glutamato descarboxilasa se halla en interneuronas, riñón, hígado, páncreas, ganglios autónomos, epífisis e hipófisis posterior; mientras la distribución de la GABA aminotransferasa es similar a la MAO: mitocondrias, médula espinal, nervios craneales, cerebelo, células gliales y células ependimarias productoras de líquido cefalorraquídeo.
Receptores para GABA
Los receptores para GABA son de varios tipos; los Ionotrópicos (GABA-A) y los metabotrópicos (GABA-B y GABA-C).El Receptor GABA-A
Situado en la membrana plasmática del terminal post sináptico es el que se relaciona con los receptores de las BZD. Por su parte los receptores GABA-B y GABA-C ubicados en la membrana plasmática de los terminales pre y post sinápticos no tienen relación con los receptores benzodiazepínicos.Los receptores GABA-A abren canales de cloro y son por lo tanto inhibidores de la conducción del impulso nervioso. Los receptores GABA-B es la permeabilidad al K+ la que aumenta, transmiten la señal por medio de segundos mensajeros. Están asociados a proteínas G. En ambas instancias el efecto es el mismo: la diferencia del potencial entre el lado interno y externo de la neurona postsináptica se incrementa, y así la célula se vuelve menos propensa a "disparar".
Aunque GABA reconoce ambos tipos de receptores, existen agonistas de GABA que sólo reconocen uno de los dos. Este hecho permitió diferenciar los dos tipos de receptores para GABA. Por ejemplo; el baclofén (Beta-p-Cloro fenil GABA), un análogo del; GABA, es inactivo en los receptores GABA-A, pero activo en los receptores GABA-B.
Los receptores GABA-A forman canales de cloro que están formados de varias subunidades. Gracias a los avances recientes en la clonación molecular, se ha logrado determinar que los receptores GABA-A contienen múltiples subunidades de receptores µ5. Asimismo, se ha sugerido que los múltiples receptores GABA-B son responsables de varias funciones metabotrópicas en el cerebro para la transmisión inhibitoria gracias a su acoplamiento con proteínas de unión GTP.
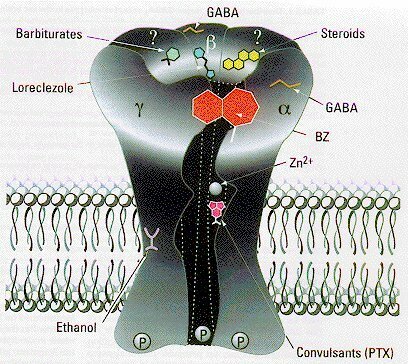
Subunidades de los receptores GABA-A
Los efectos sedante y ansiolítico de las benzodiazepinas son mediados por receptores GABAA con distinta composición de subunidades, por lo que se busca el desarrollo de fármacos que actúen específicamente sobre cada uno de estos subtipos de receptor. El refuerzo de la inhibición neuronal por el ácido gamaaminobutírico (GABA) es una de las estrategias más poderosas para el tratamiento de enfermedades como trastornos de la ansiedad, alteraciones del sueño, espasmos musculares y convulsiones. Los receptores GABAA son blanco de muchas drogas de amplio uso clínico, incluyendo los ligandos del sitio de benzodiazepinas, los barbituratos y los anestésicos. Uno de los principales objetivos en neurofarmacología ha sido dirigir selectivamente las drogas hacia subtipos específicos del receptor GABAA para refinar el espectro terapéutico de los fármacos actualmente disponibles y reducir sus efectos adversos. Los receptores GABAA, señalan los autores, son proteínas de membrana pentaméricas que funcionan como canales del cloro modulados por GABA. Estos receptores se distinguen claramente por sus subunidades, que en los mamíferos incluyen 7 clases distintas con múltiples variantes (a1 a a6, b1 a b3, g1 a g3, p1 a p3, delta, épsilon y tita). La mayoría de los receptores GABAA se componen de subunidades a, b y g.
Sinapsis GABAérgica.
En la glía la glucosa mitocondrial origina el ciclo de Krebs, dando orígen al shunt GABAérgico: glutamina-glutamato-GABA. El GABA actúa sobre los receptores postsinápticos de alta afinidad al sodio y los receptores de baja afinidad, abriendo los canales ionóforos de cloro e hiperpolarizando la membrana logra inhibir la estimulación postsináptica.
Además del canal iónico presenta:
- Sitio para el agonista no selectivo GABA.
- Sitio para el agonista selectivo 3APPA.
- Sitio para el antagonista no selectivo FACLOFEN.
- Sitio para el antagonista selectivo CGP35384.
Estructura del receptor GABA-B
Receptor GABA-B:
Se encuentra en la membrana plasmática tanto del terminal presináptico como del terminal postsináptico. No está emparentado con canales de cloro como el receptor GABA-A, sino que modulan canales de calcio y de potasio por una interacción con la proteina G y la adenil ciclasa. La unión de un agonista al receptor GABA-B presináptico disminuye la entrada de calcio originando de esta forma menor liberación de glutamato y de monoaminas. La unión de un agonista al receptor GABA-B postsináptico aumenta la salida de potasio al medio extracelular produciendo un potencial inhibitorio lento.
Fuentes:
Los receptores GABAB están asociados a una proteína G inhibitoria, disminuyendo los niveles intracelulares de AMP cíclico, que produce cambios en la permeabilidad de canales catiónicos, aumentando el influjo de iones potasio y el flujo al exterior de iones sodio. Los receptores GABAB se encuentran en su mayoría en membranas postsinápticas donde cumplen la función de heterorreceptores.
Se ha visto que receptores GABAB en el giro del cíngulo inhiben la transmisión dopaminérgica mediada por receptores D2. Se cree que esta interacción cumple un rol importante en la génesis de los síntomas positivos. Estudios funcionales han demostrado su activación durante episodios de alucinaciones auditivas. Además juega un papel importante en la determinación de funciones asociadas al pensamiento desorganizado, tales como la atención y la detección de errores
Fuente:
Metabolitos del GABA

Acido gamma hidroxibutírico GHB:
El receptor GHB, no comparte ninguna homología de secuencia con el GABA B, causa un efecto estimulante seguido por convulsiones a dosis más altas, que se cree es mediado a través del aumento de Na +/K + y liberación de dopamina y glutamato. Efectos La activación de ambos tipos de receptores, el específico del GHB y el del GABAB es responsable de sus efectos sedantes, somníferos y placenteros. Los efectos en el organismo dependerán de la dosis, ya que dependiendo de la concentración se activarán distintos receptores. Los efectos del GHB sobre la liberación de dopamina es bifásica,[30] concentraciones bajas de GHB estimulan el receptor específico estimulándose la liberación de dopamina.[31] Concentraciones más altas activan los receptores GABAB que inhiben la liberación de dopamina como hacen otras sustancias que sustituyen al GABA como el baclofen y phenibut.[32] Incrementa los niveles de la hormona del crecimiento, la prolactina y la corticosterona, aumentando la masa muscular.
Farmacocinética
Se metaboliza a nivel del citoplasma y las mitocondrias, rápidamente se convierte por transaminación en semialdehído succínico (SSA) que pasa luego a ácido succínico degradándose totalmente en el ciclo de Krebs a CO2 y agua, que se eliminarán por vía respiratoria. El semialdehído succínico se reduce en GHB: GABA → SSA → GHB.15 El GHB puede ser sintetizado directamente del GABA sin el intermediario SSA y, en una reacción inversa, el SSA puede ser sintetizado del GHB: GABA → GHB → SSA.
Acido gamma amino beta hidroxibutírico GABOB:
Se relaciona con el GABA-A receptor, inhibe también la recaptación del GABA y el GHB receptor
Sedante-Anticonvulsivo en relación al receptor GABA-A: El GABOB es un hidroxiderivado del GABA, con una acción anticonvulsivante de 5 a 10 veces superior a la del GABA. Útil en varios trastornos neurológicos, incluso como coadyuvante en el tratamiento de la epilepsia.
Estimulante suave en relación al receptor GHB: Análogo al GHB, pero no tiene efectos inductores de sueño, estimula en menor medida la hormona de crecimiento o HGH, cortisol y prolactina.
Diagrama del mecanismo de acción del neurotransmisor natural GABA (ácido gamma-aminobutírico) y de las benzodiacepinas en las células del sistema nervioso (neuronas) en el cerebro
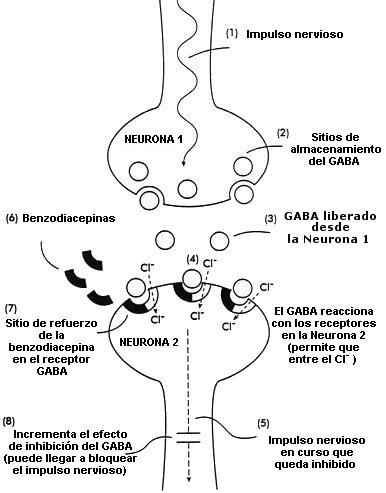
(1,2) Impulso nervioso que hace que el GABA sea liberado de los sitios en que está almacenado en la neurona 1
(3) El GABA liberado en el espacio interneuronal
(4) El GABA reacciona con los receptores de la neurona 2; la reacción permite la entrada de los iones de cloruro (Cl-) en la neurona
(5) Este efecto inhibe o detiene el progreso del impulso nervioso
(6,7) Las benzodiacepinas reaccionan con el sitio de refuerzo de los receptores GABA
(8) Esta acción aumenta los efectos inhibidores del GABA; el impulso nervioso en curso puede quedar bloqueado completamente
TODO SOBRE LAS BENZODIACEPINAS
Benzodiacepinas: Adinazolam
Farmacodinamia
Facilitan la transmisión gabaérgica y disminuyen el recambio de algunos neurotransmisores como noradrenalina, serotonina, acetilcolina y dopamina, lo que contribuye a su efecto sedativo y ansiolítico (Harvey, 1985). Al incrementar la actividad del receptor de BZD (sitio-w), estrechamente en contacto con el complejo iónico GABAA, permiten una mayor activación de los canales de Cl- por el GABA o sus agonistas (muscimol, p.ej.), permitiendo que el ion fluya al interior de la membrana, inhibiendo la excitabilidad neuronal. También se ha sugerido un incremento en la concentración de Ca++ intraneuronal dependiente de la conductancia de K+ (Tallman et al., 1980). La modulación del complejo iónico GABAA por parte de las BZD provocan cambios en la actividad eléctrica cerebral: en la vigilia, disminuyen las ondas alfa e incrementan las delta (efecto hipnótico) y las beta (principalmente en áreas frontal y rolándica (diferente a los barbitúricos). En el sueño, incrementan la actividad beta, disminuyen la latencia de inicio y el número de despertares, disminuyen las fases 1, 3, 4 y la cantidad total de sueño REM e incrementan la fase 2 y el número de ciclos de sueño REM. Al ser suspendidas abruptamente pueden llevar a un efecto de rebote con incremento de la fase REM y presentación de pesadillas y sueños extremadamente bizarros (Ballenger, 1995).
Los receptores de BZD han sido clasificados por estos dos tipos principales: los ω 1, con alta afinidad por las betacarbolinas (péptidos endógenos) y las triazolopiridazinas como Quazepam, Halazepam y Zolpidem. Son los más comunes en el cerebelo y corteza cerebral y participan en la mediación del sueño. Los ω 2 influyen en la cognición, memoria y control motor. Se hallan principalmente a nivel de la corteza (acción anticonvulsivante), hipocampo y amígdala (acción ansiolítica), y en menor cantidad en el tálamo y base del cerebro (acción sedativa). Además actúan sobre los receptores presentes en el eje hipotálamo-hipófiso-adrenal, inhibiendo la secreción de ACTH, cortisol, TSH y prolactina, que se liberan en respuesta al estrés. En el cerebelo conducen a ataxia y relajación muscular (también por efecto medular); y en el procencéfalo e hipocampo tienen efectos sobre la memoria (Zorumski & Isenberg, 1991). Últimamente se ha estado trabajando en el desarrollo de agonistas parciales que posean efectos ansiolíticos, pero sin sedación ni síndrome de abstinencia como es el caso del Abecarnil (Ballenger, 1991).
En el SNC del mamífero se han identificado por medios farmacológicos, dos subtipos de receptores ω para las benzodiazepinas. Los receptores benzodiazepina ω 1 (o BZ1)son sensibles a las β-carbolinas, a las imidazopiridinas (p. ej., zolpidem) y a las triazolopiridazinas. Los receptores ω 2 (o BZ2) de las benzodiazepinas muestran una afinidad baja por estos ligandos al tiempo que su afinidad por las benzodiazepinas es intensa. Los receptores benzodiazepina ω 1 son abundantes en el cerebelo. mientras que los receptores ω 2 son más abundantes en la médula espinal; en la corteza cerebral y el hipocampo se demuestra la presencia de ambos subtipos de receptor. Los subtipos de receptor benzodiazepina ω 1 y ω 2 también se localizan periféricamente en las células cromafines suprarrenales. Se ha identificado otro subtipo de receptor benzodiazepina, el receptor ω 3, que generalmente se denomina subtipo de receptor benzodiazepina periférico debido a su distribución en las membranas de las células gliales en tejidos extraneurales como la glándula suprarrenal, el testículo, el hígado, el corazón y el riñón. Este subtipo se detectó posteriormente en el SNC, especialmente en la membrana mitocondrial y sin relación con los receptores GABA (Gavish et al., 1992). El subtipo de receptor ω 3 presenta una afinidad elevada por las benzodiazepinas y las carboxamidas isoquinolina (Awad y Gavish, 1987). Se desconoce el papel funcional que desempeña este receptor, pero podría estar implicado en la biosíntesis de algunos esteroides neuroactivos (pregnenolona, DHEA [deshidroepiandrosterona, alo pregnenolona, tetrahidrodesoxicorticosterona) así como en la mediación «de los efectos sedantes-hipnóticos que inducen (Edgar et al., 1997; Friess et al., 1996; Ruppnecht et al., 1996).
Fuente: Tratado de psicofarmacología. Escrito por Alan F. Schatzberg,Charles B. Nemeroff.
Se considera la hipótesis de que la modulación de la subunidad del complejo macromolecular de canal de cloro del GABAa es responsable de las propiedades sedantes, anticonvulsivantes, ansiolíticas y miorrelajantes de las benzodiacepinas. El principal sitio modulador del complejo receptor GABAa está localizado en su subunidad alfa (α) y se denomina receptor de las benzodiacepinas (BZ) o receptor omega (ω). Se han identificado al menos tres subtipos del receptor (ω). Las benzodiacepinas, barbitúricos y otros fármacos con propiedades hipnóticas conocidas, interactúan con el complejo receptor GABA-BZ. A diferencia de las benzodiacepinas, que se unen no selectivamente y activan todos los subtipos del receptor omega, otros fármacos como el zolpidem se unen in vitro con el receptor (ω1) preferentemente, con un alto índice de afinidad de las subunidades alfa1/alfa5. El receptor (ω1) se encuentra principalmente en la lámina IV de las regiones sensorio motoras corticales, la sustancia nigra (pars reticulata), la capa molecular del cerebelo, el bulbo olfatorio, el complejo talámico ventral, la protuberancia, el colículo inferior y el globo pálido. Esta unión selectiva de zolpidem al receptor (ω1) no es absoluta, pero puede explicar la ausencia relativa de los efectos miorrelajantes y anticonvulsivante en estudios en animales, así como la preservación del sueño profundo (estadios 3 y 4) en estudios en humanos de zolpidem a dosis hipnóticas.
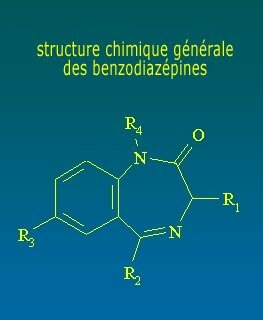
Las BZD son agentes depresores del sistema nervioso más selectivos que drogas como los barbitúricos. Actúan, en particular, sobre el sistema límbico. Las BZD comparten estructura química similar y tienen gran afinidad con el complejo de receptores benzodiazepínicos en el sistema nervioso central. Estructuralmente, las BZD presentan un anillo de benceno con seis elementos, unido a otro anillo de diazepina con siete elementos. Cada BZD específica surgirá por sustitución de radicales en diferentes posiciones.
En cuanto a los receptores específicos en el SNC para las BZD, éstos forman parte del complejo ácido gamma-aminobutírico o GABA. El GABA es un neurotransmisor con acción inhibitoria, y sus receptores forman parte de un sistema bidireccional inhibitorio conectado entre diversas áreas del SNC. Las BZD potencian la acción inhibitoria mediada por el GABA. Los receptores de las BZD se distribuyen por todo el cerebro y la médula espinal; también se encuentran en las glándulas adrenales, riñones, glándula pineal y plaquetas.
Las bdz ejercen su efecto mediante la estimulación del complejo receptor GABAa (ácido gamma-aminobutírico) lo que incrementa la entrada de cloro en la neurona disminuyendo su excitabilidad. Se han identificado varios tipos de receptores benzodiazepínicos (omega 1,2,3). Esta acción también produce un efecto inhibitorio sobre otros dos sistemas de neurotransmisión cerebral: el dopaminérgico y noradrenérgico, importantes en relación con su efectividad como tratamiento de la ansiedad. Asimismo, las bdz producen supresión de actividad en la corteza límbica y otras áreas cerebrales implicadas en la ansiogénesis.
Las propiedades farmacocinéticas de las bdz son en muchas ocasiones la base de su selección para las distintas indicaciones. Hay grandes diferencias en cuanto a su potencia, por lo que para conseguir dosis equivalentes de algunas bdz se debe multiplicar hasta por 20. También se presentan diferencias en la potencia de los efectos por separado, debido seguramente a diferencias en la afinidad por los diferentes subtipos de receptores. Por ello, algunas bdz son más efectivas que otras como anticonvulsivantes, aunque la acción ansiolítica e hipnótica deba realmente considerarse variaciones de intensidad de una misma acción farmacológica. (3)
El ratio de penetración en cerebro de estos fármacos es diferente: oxazepam lo hace relativamente despacio y por tanto tiene un comienzo de acción más lento que por ejemplo diazepam. Sin embargo, oxazepam es menos efectivo como hipnótico, pero también de menor potencial de abuso.
Pero lo que diferencia marcadamente a este grupo de medicamentos entre sí es la velocidad de eliminación. Sus vidas medias de eliminación - t1/2- varían en un amplio rango (de 2 a 100 horas) y algunos presentan metabolitos activos. La posibilidad de efectos residuales tras dosis únicas y efectos acumulativos tras administración de dosis múltiples debe considerarse, especialmente en pacientes ancianos. Las bdz potentes con tt/2 relativamente corto (triazolam, alprazolam, lorazepam) parecen causar un mayor riesgo de dependencia. Existen numerosas bdz comercializadas en nuestro país que se clasifican en función de su semivida de eliminación. En la TABLA I, (verlo más abajo)se muestra esta clasificación y se indica el tiempo de inicio del efecto, la existencia/no de metabolitos activos, las dosis usuales con el máximo recomendado, y las equivalencias en mg entre distintos principios activos para su sustitución.
Fuentes:
De Acción Prolongada t1/2: (>24 h):
De Acción Intermedia t1/2: (6-24 h): Adinazolam
De Acción Corta t1/2: (<6 h):
Ansiolíticos en asociación con Antipsicóticos:
Neurosis orgánicas o funcionales con elevado grado de ansiedad, también suelen ser usados como antidepresivos psicolépticos.
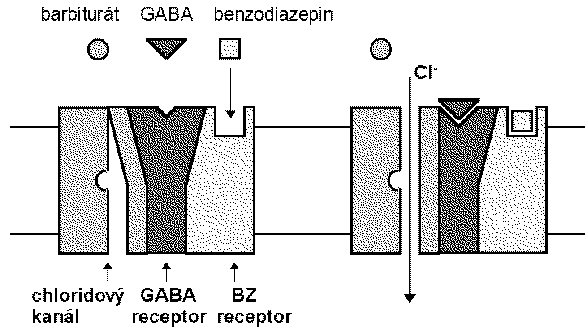
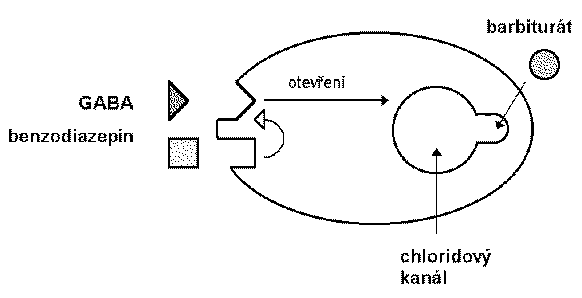
Las bzd, los barbitúricos y los nuevos sedantes cómo el zolpidem se une al canal de cloro que funciona en el receptor gaba a.
El ácido gamma-aminobutírico es el neurotransmisor inhibidor de mayor proporción en el snc. estudios eléctricos concluyen que las bzd potencian la transmisión de las neuronas gabaérgicas, incluyendo la médula espinal, corteza del cerebelo, hipotálamo, hipocampo, substancia nigra etc. las bzd incrementan la eficacia de la inhibición gabaergica en la sinapsis (vía hiperpolarización de inhibición gabaergica en la sinapsis ( vía hiperpolarización de membrana), las bzd no substituyen el gaba, más bien incrementan los efectos gaba sobre la activación del receptor asociado con el canal de cloro.
Los barbitúricos también incrementan la acción del gaba en múltiples sitios del snc, pero a diferencia de las bzd la apertura del canal dura más tiempo, inclusive a dosis altas los barbitúricos pueden tener efectos gaba-miméticos.
Fuente:
Otro:
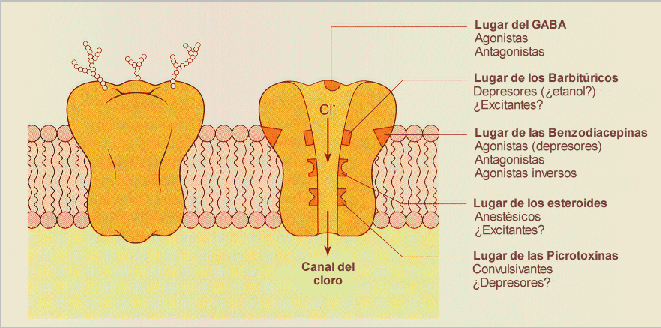
Se distinguen dos tipos de receptores GABAérgicos: el GABAA, postsináptico, y el GABAB, presináptico. El receptor GABAA es una estructura compleja que incluye al receptor GABAérgico propiamente dicho, al receptor endógeno de las benzodiacepinas y el canal iónico que, como neurotransmisor inhibitorio, es un canal de cloro (Cl-), así como la GABA-modulina, una proteína de enlace entre las estructuras principales, es decir, ente el receptor GABA y el receptor benzodiacepínico. La GABA-modulina bloquea inicialmente a los receptores e inhibe el canal iónico de Cl-; cuando esta proteína deja de actuar, ambos receptores se complementan abriendo el canal del Cl-; si alguna benzodiacepina (BZ) actúa sobre los receptores, se produce un incremento en la capacidad receptiva del propio GABA-receptor.
Fuente:
Análogos
Receptor GABA-BZD de una manera diferente a la de las benzodiacepinas:
Otros grupos surgen de la obtención de varios compuestos capaces de relacionarse con el receptor GABA-BZD de una manera diferente a la de las benzodiacepinas, dando lugar a acciones farmacológicas complejas, algunas de ellas con un posible uso terapeútico.
Ciclopirrolonas:
Farmacodinamia Agonistas del receptor gaba-benzodiacepina, con acciones farmacológicas tales como ansiolisis, relajación muscular, anticonvulsiva e hipnótica.
Imidazopiridinas:
Farmacodinamia Agonistas parciales selectivos del receptor GABA-BZD, se usan para el tratamiento corto del insomnio
Pirazolopirimidinas:
Farmacodinamia Actúan modulando la subunidad del complejo macromolecular de los canales de cloro del receptor GABA-BZD
El grupo comprende otros compuestos, completamente novedoso en su mecanismo de actuación en el tratamiento ansiolítico y que implican a otro sistema de neurotransmisión como es el de la serotonina. Actualmente se sabe que la disminución del tono serotoninérgico por diferentes medios farmacológicos conduce a un efecto ansiolítico. Se han ensayado diferentes compuestos psicotrópicos con mejores o peores resultados. Si bien es cierto que estos nuevos ansiolíticos están desprovistos de muchos de los efectos indeseables asociados a las benzodiacepinas como sedación, amnesia y farmacodependencia, no es menos evidente que no han alcanzado en muchos casos un techo terapeútico similar al de éstas. La latencia en la respuesta ansiolítica ha sido uno de las causas que ha limitado la generalización del uso de este grupo farmacológico integrado por diferentes subfamilias entre las que cabe reseñar la siguiente.
Azapironas:
Farmacodinamia Las azapironas son una clase de agentes psicoterapéuticos relativamente nueva, con propiedades ansiolíticas y antidepresivas. La buspirona es actualmente la única azapirona en uso clínico.
Su mecanismo de acción se debería al efecto agonista parcial sobre los receptores serotoninérgicos 5-HT1A.
Propiedades farmacológicas: Producen disminución del disparo de las neuronas serotoninérgicas y disminución de la síntesis y descarga de serotonina por su acción sobre ciertos receptores serotononérgicos; también tienen interacciones moderadas con los sistemas dopaminérgico y noradrenérgico cerebrales.
Farmacocinética de Benzodiacepinas y Análogos
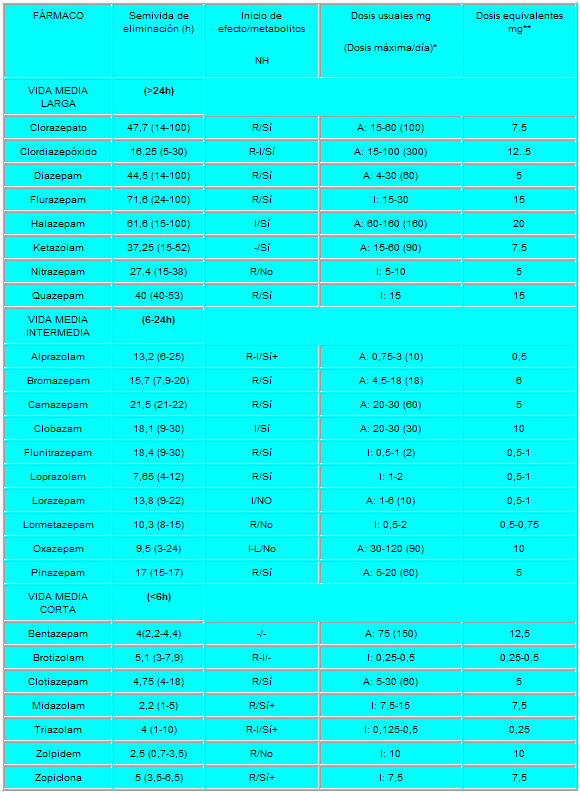
R-rápido I-intermedio L-lento NH- metabolitos activos a nivel hepático
A= ansiedad I= Insomnio+ contribuyen poco en la actividad farmacológica.
* dosis diaria en adultos vía oral en su principal indicación.
** dosis equivalentes por vía oral.
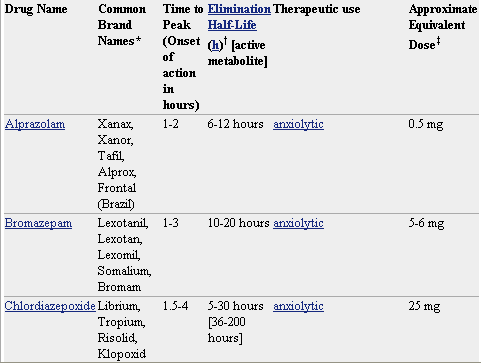

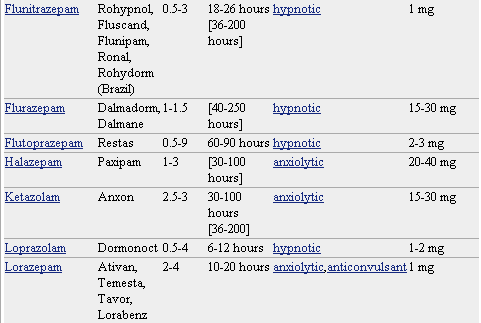

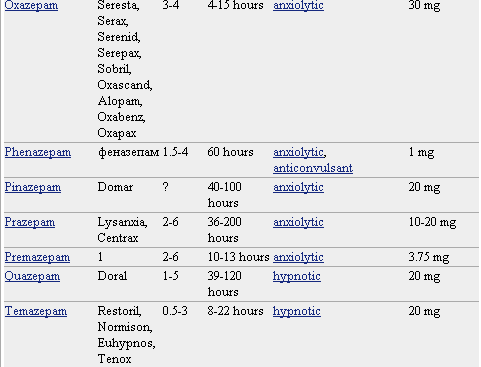
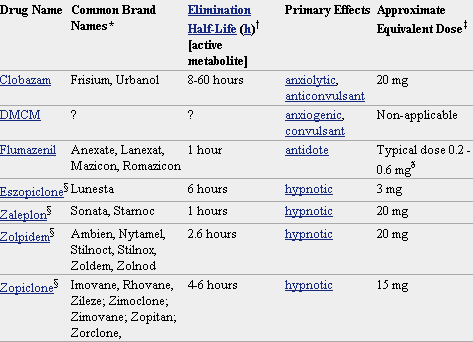
* No todos los nombres comerciales están en la lista. Haga clic en el nombre del medicamento para ver una lista más completa.
† La duración de la acción aparente es generalmente mucho menor que la vida media.Con la mayoría de las benzodiazepinas, los efectos perceptibles por lo general desaparecen después de unas horas. Sin embargo, mientras la droga está presente, tendrá efectos sutiles en el cuerpo. Estos efectos pueden hacerse evidentes durante el uso continuo o puede aparecer como síntomas de abstinencia cuando se reduce la dosis o el medicamento se deja de tomar.
‡ dosis equivalentes se basan en la experiencia clínica, pero puede variar entre los individuos. Las equivalencias se comparan con 10mg de diazepam.
§ La estructura molecular de estos fármacos difiere de la molécula de las benzodiazepinas, pero trabajan en los receptores de las benzodiazepinas con los mismos efectos o similares y se cruzan con otras drogas tolerantes.
ð El flumazenil revierte los efectos de las benzodiazepinas y otros medicamentos similares, el rango de dosis mencionadas puede variar dependiendo si la droga está siendo contrarrestado, cuál es la dosis de la droga primera, y si el flumazenil se da para realmente invertir la sobredosis o simplemente para reducir los efectos secundarios.

FARMACOCINETICA Y FARMACODINAMIA
La absorción por vía oral es buena, alcanzando concentracciones máximas entre 1 y 4 horas después de la administracción. Todas, excepto el cloracepato, se absorben inmodificadas por el tubo digestivo. La biodisponibilidad es casi completa (entre un 80-100%). Debido a su liposolubilidad, la absorción intramuscular es lenta y errática, con excepción del midazolán, cloracepán y loracepán. Por el mismo motivo, la absorción por mucosas tampoco es recomendable excepto en niños tratados con diacepán. La perfusión endovenosa ha de ser lenta y con precauciones dado el riesgo de depresión del SNC que entraña.
Se distribuyen ámpliamente por todos los tejidos, atravesando la barrera hematoencefálica y la placenta, pudiendo hallarse en la leche materna. El volumen de disponibilidad depende de la liposolubilidad relativa y se considera mayor en mujeres y población geriátrica. Tienden a acumularse en tejido cerebral y en tejidos grasos. Se unen en gran proporción a proteínas plasmáticas (70-90%), dificultándose la eliminación por diuresis forzada en el caso de las intoxicaciones agudas.
Se metabolizan extesamente sobre todo por enzimas microsomales hepáticos, originándose metabolitos que generalmente presentan mayor vida media y actividad biológica. Esta biotransformación va desde la oxidación hasta la conjugación, determinando la vida media del compuesto, que no se corresponde con la duración del efecto y constituye uno de los distintos criterios usados para clasificar estos fármacos. Este dato, junto con otras características farmacocinéticas de las principales benzodiacepinas aparecen reflejadas en la Tabla 1. Así, según la vida media de eliminación, se dividen en benzodiacepinas de acción ultracorta (vida media inferior a 5 horas), las de acción corta (entre 5 y 20 horas), acción intermedia (20 y 48 horas) y de acción larga (vida media de eliminación superior a 40 horas)
Los metabolitos conjugados son eliminados principalmente por la orina, aunque algunos compuestos sufren un proceso de circulación enterohepática, como es el caso del diacepán. Aproximadamente un 60-80% de las dosis eliminadas lo hacen por la orina y un 10% por heces, aunque este porcentaje puede variar como ya hemos comentado con anterioridad. En los últimos años, el conocimiento biológico del mecanismo de acción de las benzodiacepinas ha alcanzado un gran desarrollo. Su función se establece a partir de unos receptores específicos asociados con los sitios de unión del ácido gamma-aminobutírico (GABA), y de los canales de cloro, de tal manera que potencian los efectos inhibitorios del GABA y producen modificaciones en el resto de sistemas de neurotransmisión central. Todos los efectos benzodiacepínicos se deben a sus acciones sobre el SNC, sin modificar la actividad del sistema nervioso vegetativo ni tener acciones específicas sobre órganos aislados. Se considera, a efectos generales (con cierta variabilidad entre ellas), que las acciones más importantes son ansiolíticas, hipnóticas-sedantes, miorrelajantes y anticonvulsivantes. La acción terapéutica aparece a dosis menores que la miorrelajante y la sedante. La acción orexígena y analgésica aunque se hallan bien documentadas, no están totálmente explicadas.
EFECTOS ADVERSOS
Por norma general, son bien toleradas, presentando efectos adversos alrededor del 10% de los casos. Dichos efectos aumentan en frecuencia y gravedad cuando hay ingesta asociada de alcohol o de otros depresores del SNC.
Efectos adversos sobre SNC
La hipersedación es el más frecuente. Depende de la dosis, tiempo de administración y edad del paciente. Suele aparecer en la primera semana del tratamiento y por el fenómeno de tolerancia disminuye al final de la segunda semana. También pueden aparecer mareos, ataxia, vértigos, disartria, incoordinación, diplopia, nistagmus y rara vez parestesias. Sobre la memoria se ha constatado, por una parte, una alteración de la consolidación (relacionada con sedación y ansiolisis) y por otra parte se han descrito amnesias retrógradas (sobre todo cuando son dosis altas y la via de administración es la endovenosa). Estos efectos son más frecuentes en benzodiacepinas de vida media corta y alta potencia. En ocasiones se producen reacciones paradójicas, con base idiosincrática, caracterizadas por ansiedad, inquietud, trastornos del sueño, excitación, accesos de furia e hiperreflexia. Son más frecuentes en las dos primeras semanas del tratamiento.
Efectos sobre aparato digestivoAparecen en menos del uno por ciento. Destacan: constipación digestiva, nauseas, sequedad de boca, sabor amargo y vómitos. También se han descrito colestasis intrahepáticas y aumentos de transaminasas por daño hepático.
Efectos cardiovasculares
Raros (hipotensión arterial y taquicardia). Se han descrito efectos vasodilatadores coronarios sin efectos clínicos sobre la insuficiencia coronaria.
S. genito-urinario
Disminución del impulso sexual y alteraciones miccionales (probáblemente por hipotonía muscular), son los más frecuentes.
Otros
Reacciones alérgicas de hipersensibilidad, manifestaciones cutáneas menores, leucopenia, conjuntivitis, diplopía, visión borrosa o fiebre.
TOXICIDAD
Cabe distinguir la toxicidad aguda de la crónica. El riesgo vital por intoxicación aguda es raro, pero aumenta si a la sobreingesta se añaden otros depresores del SNC como el alcohol. Las manifestaciones clínicas suelen ser la continuación de sus efectos terapéuticos y adversos. La etiología suicida es la más frecuente, siendo las benzodiacepinas los psicofármacos más utilizados en intentos de autolisis. Las dosis tóxicas son variables. Como en el resto de intoxicaciones el tratamiento se ocupa de distintos aspectos, que van desde la eliminación del tóxico hasta la aplicación del antídoto (flumacenil)
El flumacenil, tiene afinidad por el receptor benzodiacepínico, pero carece de actividad, comportándose como un antagonista. Es soluble en agua; a diferencia del resto de las BZD es escasamente lipofílico y se une en bajo porcentaje a las proteínas plasmáticas ( 54-64% ). Su comienzo de acción es rápido, teniendo una vida media de eliminación de 1 hora. Se metaboliza en el hígado y presenta el mayor aclaramiento de todas la BZD. Ejerce su efecto al antagonizar la acción de otras BZD, por lo que su administración de forma aislada (sin la acción de otras BZD) no muestra efectos fisiológicos
Fuente:
y sin olvidarse del tratamiento sintomático de todas aquellas compliciones que puedan aparecer. Respecto a la toxicidad crónica es importante conocer el cuadro clínico producido por el uso de benzodiacepinas durante largo tiempo, sin olvidar el síndrome de abstinencia provocado tras la brusca supresión del fármaco una vez se ha producido una habituación al mismo. En ambas situaciones la tolerancia (mediada por sensibilidad receptorial) juega un papel importante. El riesgo de dependencia es bajo, siendo necesario un largo periodo de tratamiento. Dicho riesgo aumenta en pacientes con trastornos de personalidad (ansiedad crónica y síntomas disfóricos). El cuadro clínico es semejante al del uso crónico de alcohol o barbitúricos y se caracteriza por sonmolencia, vértigo, ataxia y en ocasiones nistagmus.
La supresión brusca del tratamiento, en pacientes que han desarrollado dependencia y tolerancia, puede provocar un síndrome de abstinencia más grave incluso que el de los opiáceos. Clínicamente se caracteriza por síntomas semejantes a los del cuadro original, y otros nuevos como hipersensibilidad a la luz y al sonido, malestar general, despersonalización, disforia, trastornos de la memoria, alteraciones de la percepción y psicosis agudas. La severidad del cuadro es variable y suele iniciarse entre dos y cuatro días después de la suspensión, desapareciendo progresivamente. Como tratamiento se ha empleado propanolol junto con tratamiento sintomático. Para evitar esta situación, la reducción progresiva de la dosificación y la sustitución por benzodiacepinas de vida media más larga son métodos preventivos eficaces.
Fuente: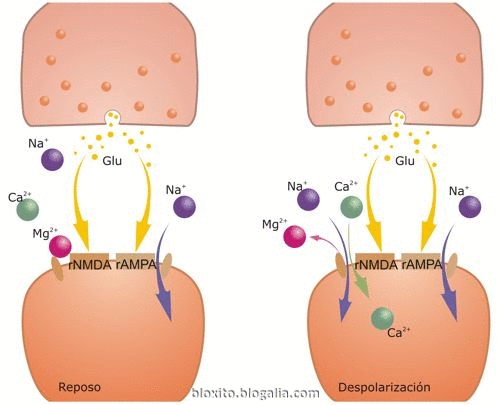
Durante la transmisión sináptica normal, el glutamato (Glu) se libera desde el botón presináptico y actúa tanto sobre receptores AMPA (rAMPA) como NMDA (rNMDA). Sin embargo, el Na+ fluye solamente a través de rAMPA, pero no de rNMDA, porque el Mg2+ bloquea el canal del rNMDA. La despolarización de la célula postsináptica libera el bloqueo del rNMDA por el Mg2+, permitiendo fluir dentro de la espina dendrítica al Na+ y al Ca2+ a través del rNMDA. El aumento resultante del Ca2+ dentro de la espina dendrítica es crítico para la puesta en marcha de la una de las formas de memoria.
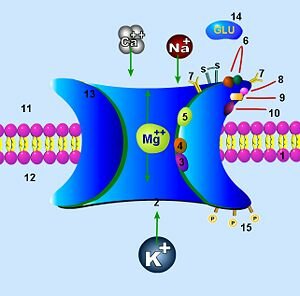
La glicina también es un co-agonista del receptor NMDA para glutamato. El número 9 señala su lugar de unión
Fuentes
![]()
Antialcohólicos Sedantes:
Reguladores equilibrio glutamato-GABA
Agonistas del GABA sedantes con apertura de los canales del cloro o reduciendo la excesiva actividad del glutamato. Tratamiento post-alcohólico, casi todos dejados de usar menos el acamprosato
Ver también antagonistas opiáceos en antialcohólicos
Entre otros:
Propanodioles como el Meprobramato, que muestra similitudes farmacológicas con los barbitúricos. Actualmente su utilización había quedado limitada al tratamiento del síndrome de abstinencia alcohólica, hasta su retirada reciente del mercado.
No es un barbitúrico. Produce farmacodependencia. Induce el metabolismo de otros fármacos. Puede provocar depresión completa, coma y muerte. Tiene absorción digestiva y tiene un metabolismo rápido. Tóxico semejante a los barbitúricos, de tipo agudo y crónico.
- Agudo: depresión progresiva, somnolencia y coma.
- Crónica: tolerancia y dependencia física y psicológica.
- No barbitúricos:
- Hidrato de Cloral
- Bromuros
- Alcohol (metanol)
- Paraldehído
Este tipo de sustancias son sedantes, pero tienen efectos secundarios muy fuertes, llegan con rapidez a provocar el coma o la muerte. Por esto aparecen los barbitúricos.
Agonistas de Receptores Melatonínicos MT1 y MT2:
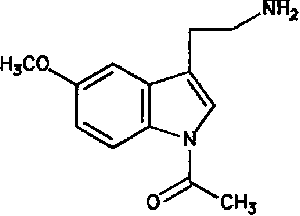
La melatonina o N-acetil-5-metoxitriptamina es una hormona encontrada en animales superiores y en algunas algas, en concentraciones que varían de acuerdo al ciclo diurno/nocturno. La melatonina es sintetizada a partir del neurotransmisor serotonina. Se produce, principalmente, en la glándula pineal, y participa en una gran variedad de procesos celulares, neuroendocrinos y neurofisiológicos. Una de las características más sobresalientes respecto a la biosíntesis pineal de melatonina es su variabilidad a lo largo del ciclo de 24 horas, y su respuesta precisa a cambios en la iluminación ambiental. Por ello, la melatonina se considera una neurohormona con función pertinente en la fisiología circadiana. La melatonina es producida por los pinealocitos en la glándula pineal (localizada en el cerebro), la cual produce la hormona bajo la influencia del núcleo supraquiasmático del hipotálamo, el cual recibe información de la retina acerca de los patrones diarios de luz y oscuridad. La glándula pineal de los humanos tiene un peso cercano a los 150 miligramos y ocupa la depresión entre el colículo superior y la parte posterior del cuerpo calloso. A pesar de la existencia de conexiones entre la glándula pineal y el cerebro, aquélla se encuentra fuera de la barrera hematoencefálica y está inervada principalmente por los nervios simpáticos que proceden de los ganglios cervicales superiores.
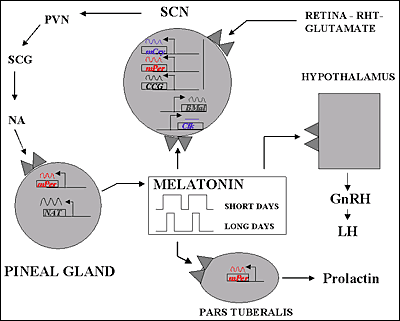
Representación esquemática del control de la producción y las funciones de la melatonina
La secreción de melatonina desde la glándula pineal queda inhibida por la luz brillante, por lo que la menor concentración de melatonina sérica se observa durante el día. Al comienzo de la noche, hay un incremento en la liberación de noradrenalina que activa los b-adrenoceptores de la glándula pineal para aumentar la formación de AMPc y con la activación de los a1-adrenoceptores se amplifica más la respuesta. Este segundo mensajero provoca la activación de la serotonina N-acetiltransferasa que va a incrementar la síntesis de melatonina. Existen dos subtipos de receptores de melatonina, MT1 y MT2, ambos están acoplados a proteínas Gi/o. Por medio de la utilización de receptores recombinantes se ha observado que MT1, por su actuación por medio de proteínas G produce la inhibición de la adenilato ciclasa y, por otra parte, produce la activación de la Fosfolipasa C. Mientras que MT2, además dela inhibición de la adenilato ciclasa produce la inhibición de la ruta de la guanidil ciclasa. Todo ello sucede una vez que la melatonina se una a sus receptores. Por tanto, la glándula pineal funciona como un transductor neuroendocrino. En mamíferos, la información fotosensorial que entra por la retina influye en la actividad de sus proyecciones neuronales, que sirve finalmente para inhibir o estimular la secreción de serotonina. En animales aislados en oscuridad continua el ritmo circadiano de secreción de melatonina. La síntesis de melatonina se lleva a cabo desde un reloj endógeno, probablemente situado dentro del núcleo supraquiasmático del hipotálamo, habiendo sido entrenado normalmente en el ciclo día-noche.
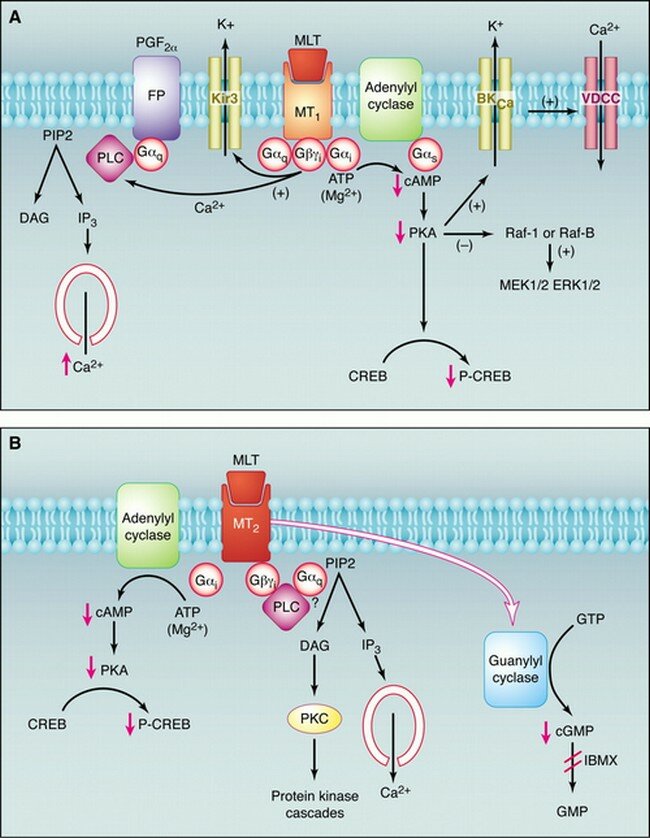
Pinchar en la imagen para agrandar
En mamíferos, los receptores de melatonina se encuentran en el encéfalo y en algunos órganos periféricos. Aunque hay una considerable variación en la densidad y localización de la expresión de receptores entre las distintas especies.El subtipo MT1 se encuentra en la pars tuberalis de la hipófisis y en el núcleo supraquiasmático del hipotálamo. El subtipo MT2 se encuentra principalmente en la retina. El subtipo Mel1C de vertebrados no mamíferos se expresa en muchas áreas cerebrales.

Ya casi no se usan sustituidas por las Benzodiazepinas
Aunque los barbitúricos constituyeron un tratamiento inicial de la ansiedad, su actividad ansiolítica es escasa y se encuentra ligada directamente a su actividad sedante. Los barbitúricos se asocian a importantes problemas de dependencia y abstinencia, y su perfil de seguridad es bajo, sobre todo cuando se asocian con otras drogas depresoras como el alcohol. Desplazados por las BZD, no se suelen usar ya como hipnótico-sedantes, aunque se siguen utilizando como antiepiléptico y anestésico general.
Son productos sintéticos que derivan del ácido barbitúrico obtenido por Bayer en 1863. El primer barbitúrico introducido en terapéutica a principios de este siglo, fue el barbital
Depende del barbitúrico empleado y de la dosis administrada.• Dosis altas de barbitúricos de acción ultracorta conducen a la anestesia. • Dosis medias de barbitúricos de acción intermedia y corta, les hacen actuarcomo sonniferos o hipnóticos. • Dosis bajas de barbitúricos de acción prolongada les hacen actuar como sedantes. En el caso de Fenobarbital no se puede descartar el efecto hipnótico a dosis bajas.Sobre el sistema nervioso central SNC.Su acción farmacologica fundamental es la depresión no selectiva, del SNC, que según dosis puede ir desde la sedación, hasta la anestesia general, como respiratorio, y aun la muerte, por parálisis del centro respiratorio ( bulbo raquídeo ).
En resumen, las características de la farmacodependencia de tipo barbitúrico son las siguientes :1.-Dependencia psíquica variable, pero a menudo marcada, relacionada con los efectos deseados de la droga.2.-Dependencia física marcada cuando las dosis son notablemente superiores a los niveles terapéuticos. La supresión brusca pone en peligro la vida si nohay tratamiento medico adecuado. 3.-La tolerancia que se establece es distinta para los distintos efectos de la droga. Hay una tolerancia cruzada, mutua pero incompleta, entre el alcohol y los barbitúricos.
Los Barbitúricos pertenecen a un grupo de medicamentos llamados depresores del Sistema Nervioso Central (SNC). Pueden actuar tanto en el cerebro como en el SNC produciendo efectos que pueden ser tanto positivos como dañinos. Esto depende de la condición individual de cada persona y su respuesta a la dosis de la medicina tomada. Algunos de los barbitúricos pueden ser usados antes de una cirugía para aliviar la ansiedad o tensión. Además algunos de estos son usados como anticonvulsivos para ayudar a controlar algunos síntomas tales como la epilepsia.
También han sido utilizados para tratar el insomnio; pero si son usados con regularidad, no serán efectivos luego de las dos semanas de toma consecutiva. Los barbitúricos también han sido usados para aliviar el nerviosismo o alteración durante el día. Sin embargo, han sido reemplazado en su mayoría por medicación más segura ya que si se utiliza en grandes cantidades, o por largos períodos produce hábito.
Los Barbitúricos se encuentran dentro de las drogas más adictivas. Son generalmente un sustituto para el alcohol (ya que produce efectos similares). La gente los utiliza para obtener una sensación de euforia y relajación. De todas formas, su uso es ilegal sin prescripción y supervisión médica. Los barbitúricos fueron la primera clase de agentes sedantes-hipnóticos conocidos y fueron una vez extremadamente populares como drogas de abuso. En la actualidad, las benzodiazepinas han reemplazado ampliamente a éstos para las terapias en pacientes, lo que ha creado un baja en el abuso de barbitúricos. Otro importante factor fue el refuerzo de las leyes para la restricción de su venta.
Historia:
Los barbitúricos son productos sintéticos que derivan del ácido barbitúrico obtenido por Bayer en 1863. El primer barbitúrico introducido en terapéutica a principios de este siglo, fue el barbital.
Más de 2,500 barbitúricos han sido sintetizados, y en la cima de su popularidad, alrededor de 50 fueron llevados al mercado para uso humano. Hoy en día, alrededor de una docena siguen siendo utilizados por la medicina.
Los barbitúricos producen un amplio espectro de depresión del SNC, desde una sensación leve hasta el coma, y han sido utilizados como sedantes, hipnóticos, anestésicos, y anticonvulsivos. La principal diferencia entre muchos de estos productos, es la rapidez con la cual producen el efecto y por cuanto tiempo persiste ese efecto. Los mismos se clasifican en acción ultra corta, corta, intermedia y prolongada.
Efectos:- Efectos a corto plazo: (duran por 15 horas luego de la ingesta)
- • Alivio de la tensión y la ansiedad
- • Somnolencia
- • Sentimiento de borrachera / intoxicación
- • Discurso poco claro
- • Inhabilidad para controlar funciones corporales simples (caminar, balance, etc.)
- • Disminución de la memoria
- • Inestabilidad Emocional
-
-
- Efectos a Largo Plazo:
- • Cansancio crónico
- • Falta de coordinación general
- • Problemas de visión
- • Mareos
- • Disminución de los reflejos y respuesta
- • Disfunción sexual
- • Irregularidades menstruales
- • Desordenes respiratorios
- Tolerancia:
- • Se desarrolla muy rápidamente
- • Puede requerirse hasta 10 veces la dosis original para producir el mismo estado
- Síntomas de Abstinencia:
- • Alucinaciones
- • Desordenes de alimentación
- • Desorientación
- • Vómitos
- • Desórdenes del sueño
Barbitúricos junto a otros depresores: La combinación de barbitúricos con otras drogas esmuy peligrosa (especialmente cuando se combinan con otros depresores del SNC como el Demerol, la heroína, morfina, y codeína). Los barbitúricos producen muchos de los mismos efectos que el alcohol, y la combinación de ambos es a menudo letal. Hay un peligro oculto con el uso de los barbitúricos, especialmente si se sufre de alergias. Las Antihistaminas (encontradas en la mayoría de las medicaciones para alergias, resfrios, y sinusitis) son otro tipo de depresores del SNC, y cuando son tomados en combinación con los barbitúricos, pueden producir el fallo respiratorio.
Barbitúricos junto a estimulantes: Los barbitúricos son a menudo contrarrestados con altas dosis de anfetaminas o cocaína (ambos estimulantes). Esta mezcla es extremadamente peligrosa, ya que puede acelerar el ritmo cardíaco y producir una falla coronaria.
Patofisiología: Los barbitúricos se unen a un sitio específico en los canales iónicos sensibles al ácido gamma-aminobutírico (GABA) encontrados en el SNC, donde permiten la entrada de cloruro a las membranas celulares y, subsecuentemente, hiperpolariza la neurona postsináptica.
GABA es el mayor neurotransmisor inhibotorio del SNC. Los barbitúricos producen corrientes de cloruro mediado por el GABA al unirse al complejo receptor GABA A-ionoforo e incrementando la duración de la apertura del ionoforo; los barbitúricos inhiben la despolarización neuronal al potenciar y prolongar las acciones de GABA. En altas dosis, los barbitúricos estimulan los receptores GABA A directamente en la ausencia del GABA. Los barbitúricos también bloquean los receptores de glutamato (neurotransmisor excitatorio) en el SNC.
Los barbitúricos pueden agruparse funcionalmente en agentes de acción prolongada y corta (conformando agentes de acción ultra corta, corta, e intermedia). En comparación con agentes de acción prolongada, los de corta duración son más liposolubles, mas enlaces a proteínas, mayor pKa, mayor velocidad de acción y mas corta duración, y se metabolizan casi en su totalidad en el hígado para inactivar metabolitos (los cuales son excretados como glucurónidos en la orina). Los agente de larga acción, los cuales son menos liposolubles, se acumulan mas lentamente en los tejidos y son excretados mas rápidamente por el riñón como una droga activa. A propósito, la excresión urinaria cuenta para el 20-30% de la eliminación del fenobarbital y el 15-42% del primidone (ambos de acción prolongada).
Los agentes de corta duración tienen una eliminación de vida media de menos de 40 horas en comparación a los de acción prolongada, que tienen una vida media de eliminación mayor a 40 horas.
Los barbitúricos estimulan el sistema del citocroma hepático P-450 mezclado con la enzyme microsomal oxidasa; por lo tanto, afectan los niveles de droga de medicaciones que son dependientes de este sistema (por ej, coumadina).
Efectos al Sistema Nervioso Central
Los barbitúricos actúan principalmente en el SNC y, en consecuencia, afectan otros sistemas de órganos. Los efectos directos incluyen ser sedantes e hipnóticos en bajas dosis. Los barbitúricos lipofílicos, tales como el tiopental, causan anestesia rápida por su tendencia a penetrar en el tejido cerebral rápidamente. Los barbitúricos son todos anticonvulsivos ya que hiperpolarizan las membranas celulares; y por ende son eficaces en el tratamiento de la epilepsia.
Efectos Pulmonares Los barbitúricos pueden causar depresión del centro medular respiratorio e inducir a la depresión respiratoria. Los pacientes con enfermedades crónicas de obstrucción respiratoria (COPD) son más susceptibles a estos efectos, inclusive en dosis que serían consideradas terapéuticas en individuos sanos. La fatalidad por sobredosis es generalmente secundaria a la depresión respiratoria.
Efectos Cardiovasculares La depresión cardiovascular puede darse por la depresión de los centros medulares vasomotores; pacientes con falla congestiva cardiaca (CHF) son más susceptibles a estos efectos. En altas dosis, la contractilidad cardiaca y el tomo vascular quedan comprometidos, lo que puede resultar en el colapso cardiovascular.
Mortalidad/Morbilidad: La fatalidad asociada a la sobredosis de barbitúricos es poco común, pero abundan las complicaciones. La morbilidad incluye neumonía, shock, hipoxia, y coma.
Webliografía
Los antihistamínicos son fármacos que se usan para contrarrestar o bloquear los efectos causados en el organismo por la liberación de histamina.
La histamina es una amina primaria derivada del imidazol. Es una amina biógena que se encuentra ampliamente distribuida en las mucosas del tracto gastrointestinal y respiratorio, así como en la piel. La mayor fuente de histamina en el cuerpo humano son los mastocitos tisulares, lo que da origen a su nombre actual que deriva de la palabra griega histos = tejido. Esta se almacena en forma inactiva dentro de los gránulos basófilos de los mastocitos tisulares y leucocitos circulantes. En respuesta a ciertos estímulos, tales como un daño epitelial producido por venenos o toxinas, estas células liberan histamina, que inmediatamente produce la dilatación de los vasos sanguíneos, es decir, una reacción inflamatoria.
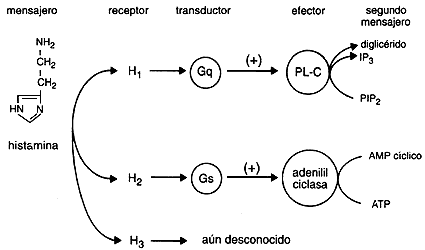
Existen al menos tres tipos distintos de receptores para la histamina (H1 - H2 - H3). También la histamina funciona como un neurotransmisor en el Sistema Nervioso Central ya que se encuentra altas concentraciones de receptores H1 en el tálamo, hipotálamo y ciertas regiones del cerebelo y cerebro anterior. Las neuronas que contienen histamina participarían en la regulación de la sed, la temperatura corporal y la secreción de hormona antidiurética, así como en el control de la presión sanguínea y la percepción del dolor.
Los Receptores H1 están relacionadas con la respuesta alérgica inmediata como: fiebre del heno, secreción nasal, estornudos, el picor de nariz y garganta, y en menor grado, las molestias de la conjuntivitis y de la dificultad respiratoria. También pueden disminuir el picor y la erupción de las alergias alimenticias.
Los Receptores H2 se encuentran involucrados en la regulación de la secreción del jugo gástrico y ácido clorhídrico estomacal, de modo que los antagonistas H2 son ampliamente utilizados en el tratamiento de la úlcera péptica.
Los Receptores H3 parecen estar presentes en las terminaciones nerviosas histaminérgicas, donde ejercen una regulación del tipo feed-back(realimentacion)
Cuando es liberada en los tejidos genera una respuesta caracterizada por:
* Secreción de serosas y mucosas.
* Estimulación de terminaciones nerviosas que provocan prurito (o dolor).
* Dilatación vascular periférica y aumento de la permeabilidad capilar
* También contrae a la musculatura lisa (como los músculos lisos bronquiales) y a las glándulas.
A estos fenómenos en conjunto se conocen como "Inflamación Alérgica".
En forma arbitraria los "antihistamínicos" han sido clasificados clínicamente de acuerdo a la capacidad depresora del SNC en:
A) Antihistamínicos Clásicos o de Primera Generación.
B) Antihistamínicos No Sedantes o de Segunda Generación.Mecanismo de acción: tanto los antihistamínicos H1, como los antihistamínicos H2 actúan como antagonistas competitivos de los receptores de la histamina llamados H1 ( ubicados de manera abundante en músculo liso de bronquios e intestino) La estimulación de estos receptores causa manifestaciones de alergia. La dilatación de los vasos sanguíneos periféricos se debe a efectos de la histamina en los receptores tanto H1, como H2. Los receptores de la histamina tipo H2 se localizan a nivel del estómago (mucosa), son los responsables de estimular las secreciones de ácido clorhídrico
Antihistamínicos tipo H1.
Efectos de loa antihistamícos tipo H1.
1.-Ación antihistamínica.-inhiben los efectos de la histamina, pues compiten por los receptores celulares para esta sustancia. Es decir a nivel de tubo digestivo inhiben la contracción muscular, a nivel de bronquios antagonizan el efecto broncoconstrictor causado por la histamina. Disminuyen la permeabilidad capilar reduciendo la formación de edema. Antagonizan los efectos de vasodilatación periférica
2.-Efectos sobre el SNC..-con frecuencia lo deprimen, lo cual se manifiesta como ataxia o laxitud, ocasionalmente producen excitación. Algunos son eficaces para prevenir mareos (pero no son útiles si el mareo ya esta presente).
3.-Acción anticolinérgica.—algunos antihistamínicos en especial los derivados de etanolamina y etilendiamina pueden antagonizar a la acetilcolina liberada en los nervios periféricos, es decir producen efectos semejantes a los de la atropina.
4.-Acción antiadrenérgica.------los antihistamínicos derivados de la fenotiacina ejercen ligero efecto de bloqueo de los receptores alfa adrenérgicos.
5.-Acción anestésica local.-------La difenhidramina y la prometacina principalmente, pero en realidad la mayoría de los antihistamínicos pueden producir anestesia local debido a que bloqquean los canales del Na en la célula.
6.-Acción antiserotoninérgica.-----Algunos antihistamínicos, por ejemplo la ciproheptidina pueden bloquear a los receptores para la serotonina.
Principales Efectos tóxicos de los antihistamínicosSedación, ataxia, si se administran vía oral pueden ocasionar anorexia, náusea, vómito, constipación y diarrea. Los antihistamínicos derivados de la piperacina (ciclina, meclicina, clorociclina) pueden tener efectos teratogénicos, deben usarse con precaución en animales gestantes. Algunos antihistamínicos aplicados tópicamente pudieran producir dermatitis alérgica
Receptores de Histamina.
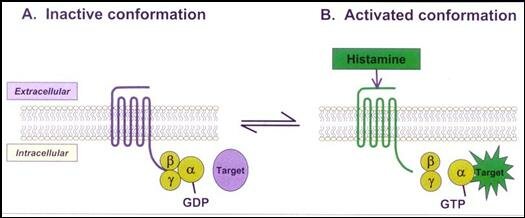
Hasta el momento actual se han encontrado 4 subtipos diferentes de receptores para Histamina en humanos (H1, H2, H3 y H4). Los 4 subtipos pertenecen a la familia de GPCRs. Son proteínas integrales de la membrana y están formados por una cadena en forma de serpentina que entra y sale 7 veces de la membrana. En su extremo extracelular se encuentra el sitio para acoplar al ligando (histamina o agonista o agonista inverso) y en el extremo intracelular se encuentra asociado a una proteína G la cual a su vez está formada por 3 subunidades (alfa, beta y gamma) las cuales se pueden asociar (abg) o disociar (a - bg) dependiendo de la actividad del receptor. Acoplado a la subunidad alfa se encuentra una molécula de GDP de baja energía que se puede fosforilar a GTP de alta energía cuando se disocia la subunidad alfa del heterodímero beta/gamma y actúa sobre una enzima “blanco”.
Receptores H1.
En el brazo corto del cromosoma 3 (3p) se encuentra codificada la información para la síntesis de estos receptores. La unión de la histamina a los receptores H1 estimula a la Fosfolipasa C (PLC) la cual a su vez cataliza a los fosfolípidos de las membranas para obtener 2dos. mensajeros: inositol trifosfato (IP3) y diacilglicerol (DAG). Estos mensajeros activan la cascada de la Protein cinasa C y la movilización del calcio intracelular permitiendo que se activen microtúbulos y microfilamentos del citoplasma para transportar gránulos con diferentes moléculas en su interior y eventualmente ser expulsados de la célula para ejercer algún efecto sobre las células vecinas. También se ha encontrado que el receptor H1 puede estar asociado a otras fosfolipasas como la PLD e inclusive la PLA-2. Recientemente también se ha encontrado que los receptores H1 pueden activar a través de los 2dos. mensajeros al Factor nuclear kB (NF-kB) el cual a su vez desregula genes en el DNA permitiendo la síntesis de varias citocinas y moléculas de adhesión para que se liberen o expresen sobre la membrana de la célula blanco. Los efectos de los receptores H1 se han relacionado fácilmente con los síntomas y signos de diferentes enfermedades alérgicas. Rinorrea, prurito, estornudos y el edema que causa obstrucción nasal en la Rinitis alérgica. El lagrimeo, la hiperemia, el prurito y la quemosis en la conjuntivitis alérgica. En el asma los receptores H1 pueden ocasionar contracción del músculo liso bronquial y estimular la producción de moco en las glándulas pero este efecto es menor comparado al de los leucotrienos sobre sus receptores. En la piel los receptores H1 ocasionan contracción de las células endoteliales de las vénulas post capilares causando la aparición de ronchas y estimulan a las fibras C para ocasionar prurito.
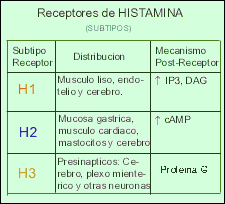
Fuentes:
Uso residual
6 meses para responder posts antiguos
¡Queremos tu opinión!
Si pregunta sobre este fármaco revise antes estos dos enlaces en particular
Ahora ya puede entender el prospecto y la ficha técnica
-